Output Files
For this example we will use the directory 03_extractions previously created with the extract module. We run the following Captus command to collect markers across samples and align them:
captus align -e 03_extractions_CAP/ -o 04_alignments_CAP -m ALL -f ALL
After the run is finished we should see a new directory called 04_alignments with the following structure and files:
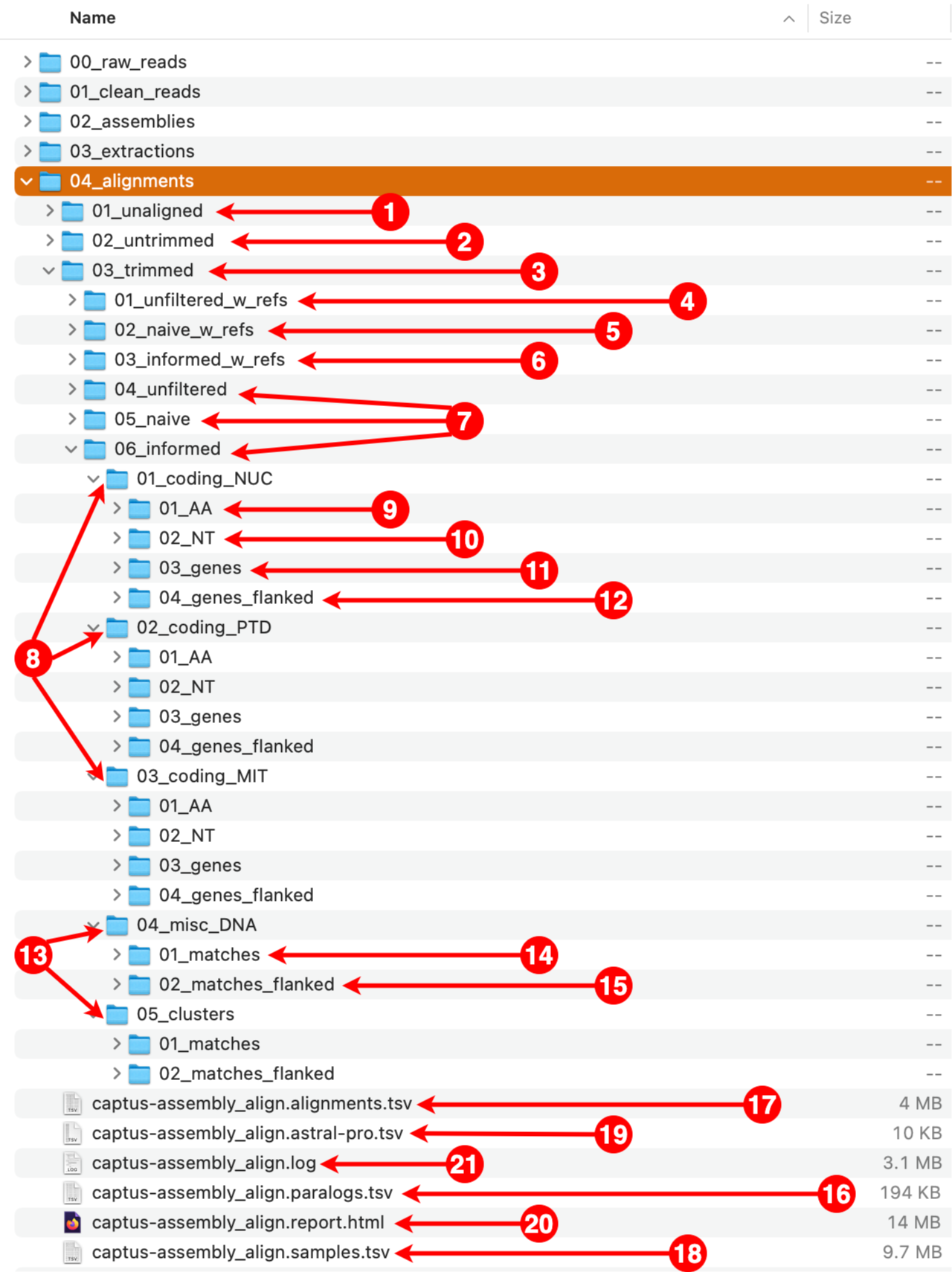

1. 01_unaligned
This directory contains the unaligned FASTA files corresponding to each marker that were gathered from the extractions directory. The files are organized in subdirectories, first by marker type and then by format.
2. 02_untrimmed
This directory contains the aligned FASTA files corresponding to each file in the 01_unaligned directory. The files are organized in subdirectories, first by filtering strategy, then by marker type, and finally by format. The subdirectory structure is identical to the one inside the 03_trimmed directory (see 4 to 15 below).

3. 03_trimmed
All the files present in the 02_untrimmed directory are trimmed using ClipKIT which removes columns that are mostly empty (see options --clipkit_algorithm, --clipkit_gaps), then Captus removes sequences that are too short after trimming (--min_coverage). The files are organized in subdirectories, first by filtering strategy, then by marker type, and finally by format. The subdirectory structure is identical to the one inside the 02_untrimmed directory (see 4 to 15 below).

4. 01_unfiltered_w_refs
This directory contains the alignments before performing any filtering. All the reference sequences selected by at least a sample will be present as well as all the paralogs per sample. The files are organized in subdirectories, first by marker type and then by format.
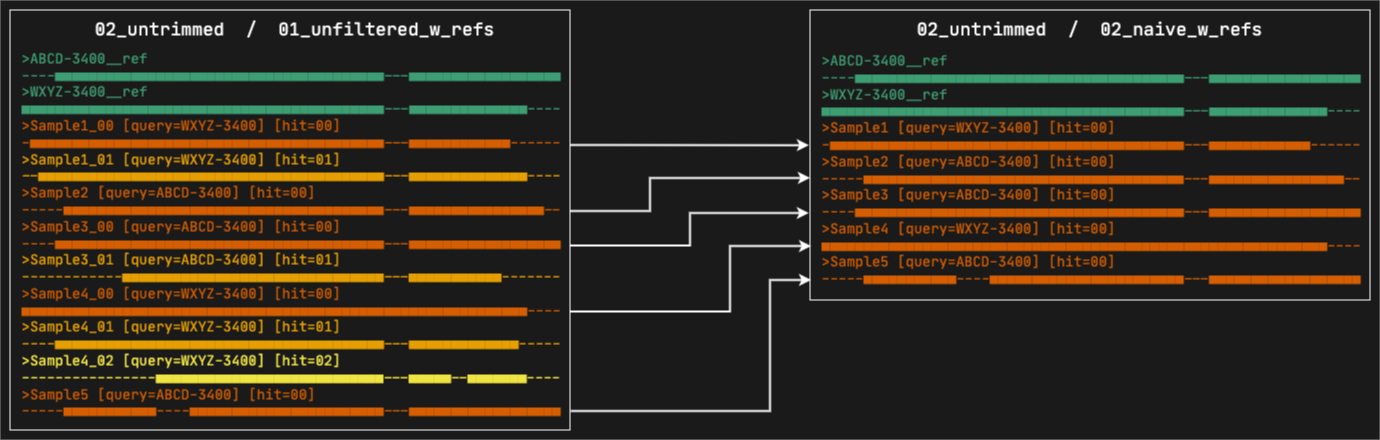
5. 02_naive_w_refs
This directory contains the alignments where paralogs have been filtered by the naive method, which consists in simply keeping the best hit per sample (hit ranked as 00). All the reference sequences selected by at least a sample will still be present. The files are organized in subdirectories, first by marker type and then by format.
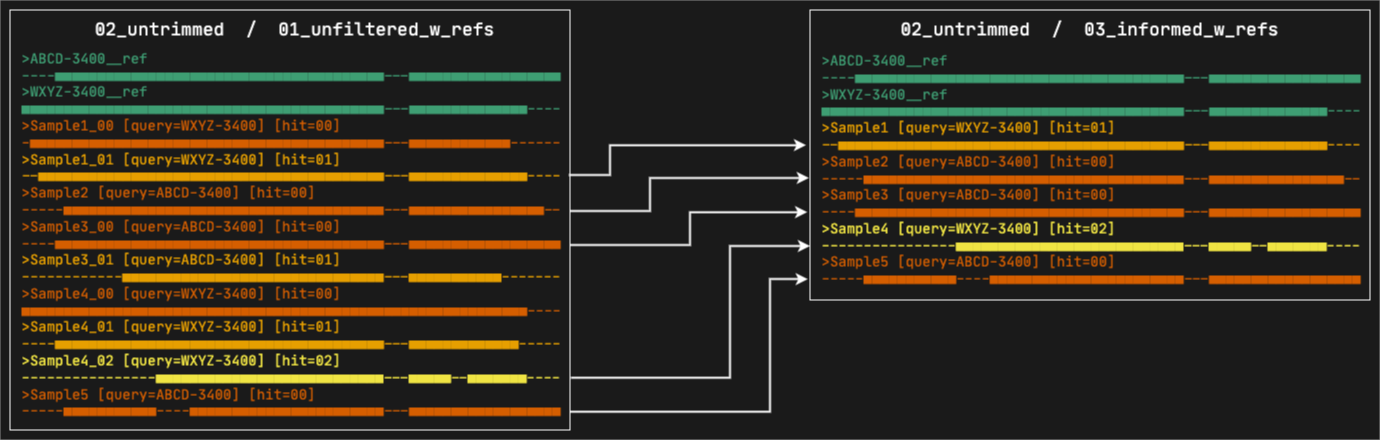
6. 03_informed_w_refs
This directory contains the alignments where paralogs have been filtered by the informed method. Under this strategy, Captus compares every copy to the most commonly used reference sequence (sequence ABCD-3400 in the figure) and retains the copy with the highest similarity to that reference, regardless of its paralog ranking (in the figure, Sample1 and Sample4 whose selected copies had paralog rankings of 01 and 02 respectively). All the reference sequences selected by at least a sample will still be present. The files are organized in subdirectories, first by marker type and then by format.
7. 04_unfiltered, 05_naive, 06_informed
These contain equivalent alignments to directories 01_unfiltered_w_refs, 02_naive_w_refs, and 03_informed_w_refs respectively, but excluding the reference sequences. In most cases you will estimate phylogenies from the trimmed versions of these alignments.
8. 01_coding_NUC, 02_coding_PTD, 03_coding_MIT
These directories contain the aligned coding markers from the NUClear, PlasTiDial, and MITochondrial genomes respectively.
The alignments are presented in four formats: protein sequence (coding_AA), coding sequence in nucleotide (coding_NT), exons and introns concatenated (genes), and the concatenation of exons and introns flanked by a fixed length of sequence (genes_flanked):
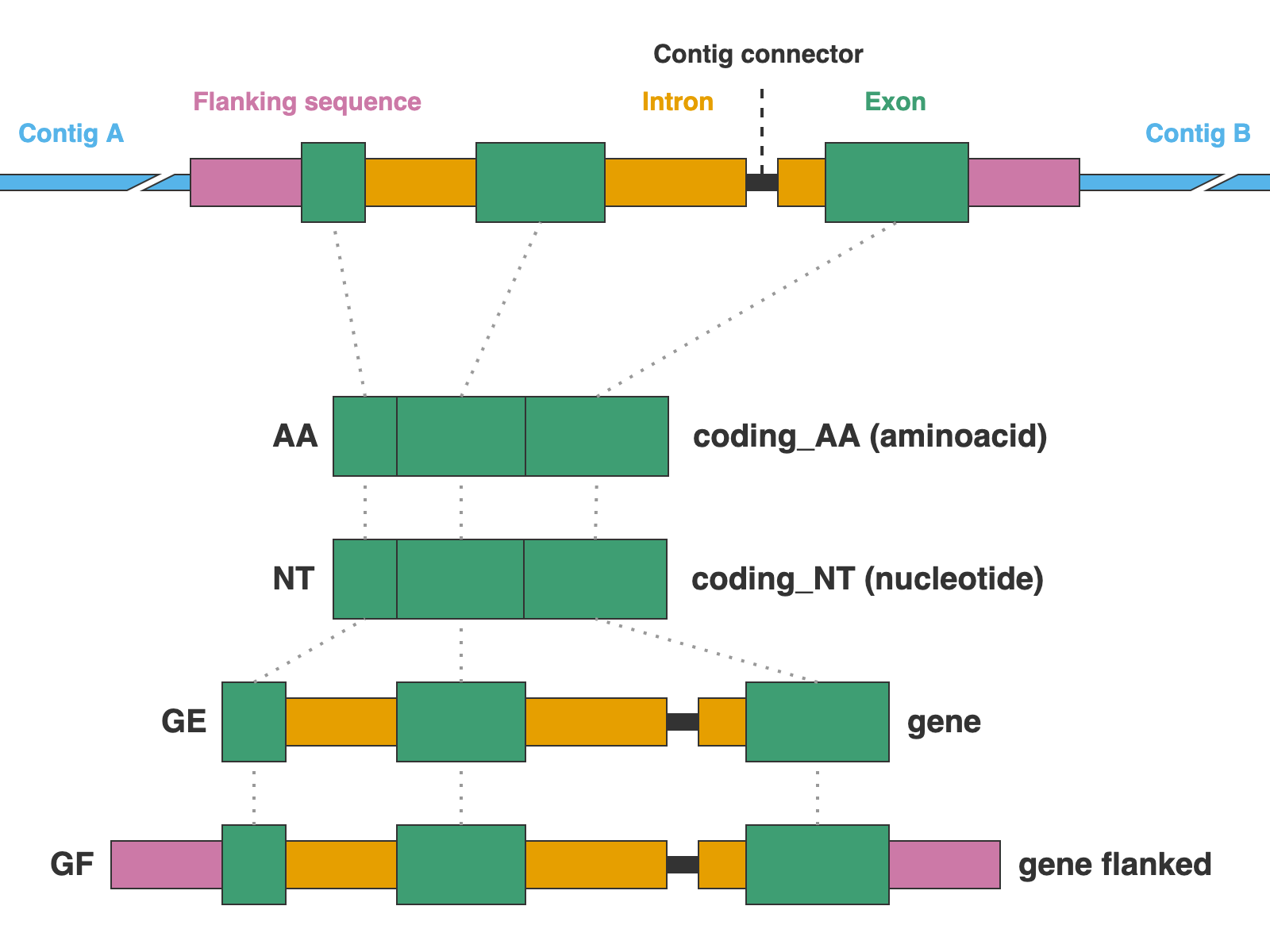
9. 01_AA
This directory contains the protein alignments (AA in the figure above) of the extracted markers gathered across samples. One FASTA file per marker, with extension .faa.
10. 02_NT
This directory contains the alignments of coding sequence in nucleotides (NT in the figure above) of the extracted markers gathered across samples. One FASTA file per marker, with extension .fna.
11. 03_genes
This directory contains the alignments of gene sequence (exons + introns) in nucleotides (GE in the figure above) of the extracted markers gathered across samples. One FASTA file per marker, with extension .fna.
12. 04_genes_flanked
This directory contains the alignments of flanked gene sequence in nucleotides (GF in the figure above) of the extracted markers gathered across samples. One FASTA file per marker, with extension .fna.
13. 04_misc_DNA, 05_clusters
These directories contain the aligned miscellaneous DNA markers, either from a DNA custom set of references or from the CLusteRing resulting from using the option --cluster_leftovers during the extraction step.
The alignments are presented in two formats: matching DNA segments (matches), and the matched segments including flanks and other intervening segments not present in the reference (matches_flanked).
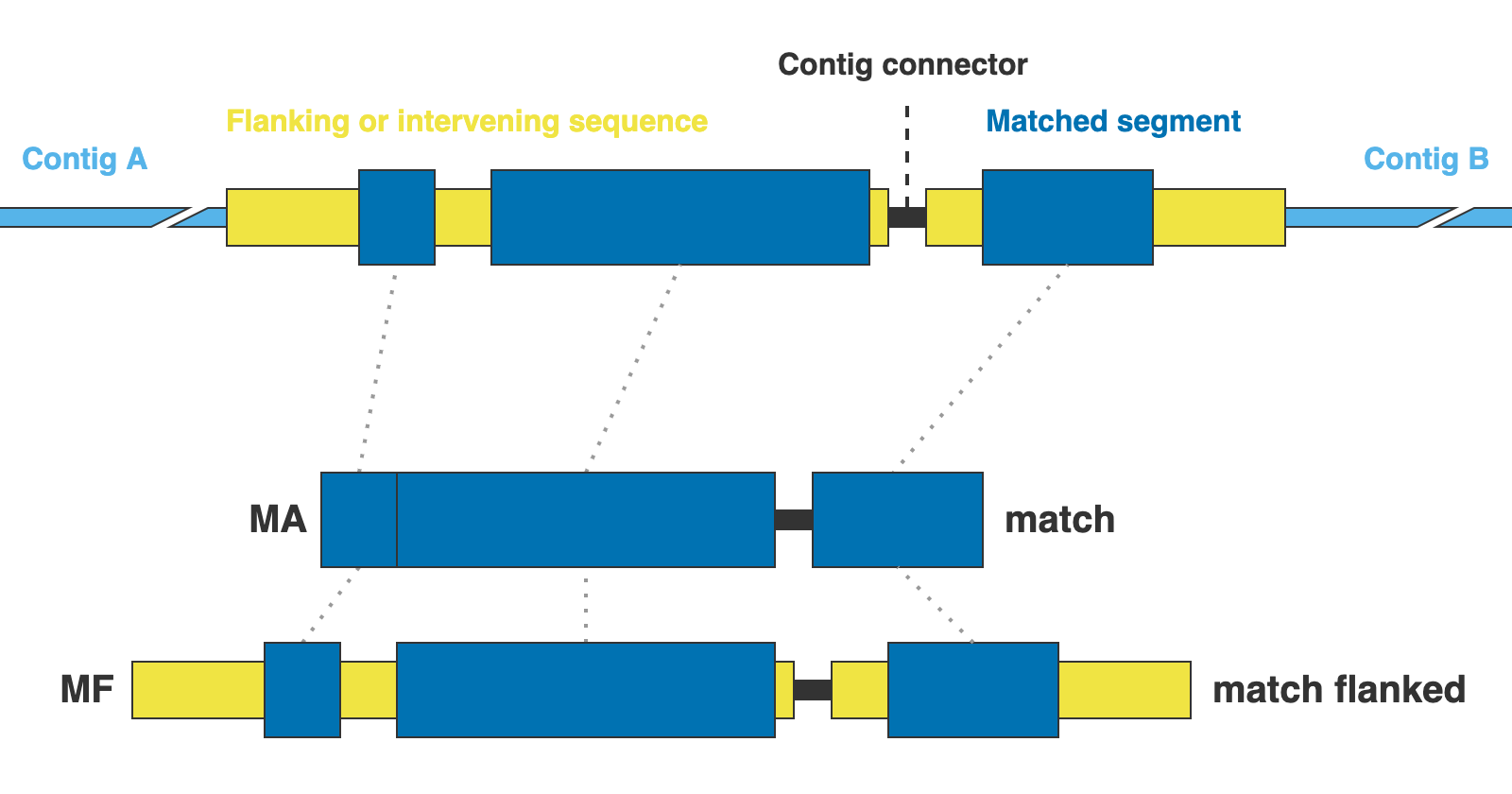
14. 01_matches
This directory contains the alignments of DNA sequence matches (MA in the figure above) for the extracted markers gathered across samples. One FASTA file per marker, with extension .fna.
15. 02_matches_flanked
This directory contains the alignments of DNA sequence matches (MF in the figure above) including flanks and intervening segments not present in the references for the extracted markers gathered across samples. One FASTA file per marker, with extension .fna.
16. captus-align_paralogs.tsv
A tab-separated-values table recording which copy was selected during the informed filtering of paralogs.
17. captus-align_alignments.tsv
A tab-separated-values table recording alignment statistics for each of the alignments produced.
18. captus-align_samples.tsv
A tab-separated-values table recording sample statistics across the different filtering and trimming stages, as well as marker types and formats.
19. captus-align_astral-pro.tsv
ASTRAL-Pro requires a tab-separated-values file for mapping the names of the paralog sequence names (first column) to the name of the sample (second column). Captus produces this file automatically.
20. captus-align_report.html
This is the final Aligment report, summarizing alignment statistics across all processing stages, marker types, and formats.
21. captus-align.log
This is the log from Captus, it contains the command used and all the information shown during the run. Even if the option --show_more was disabled, the log will contain all the extra detailed information that was hidden during the run.
Created by Edgardo M. Ortiz (06.08.2021)
Last modified by Edgardo M. Ortiz (23.12.2024)