Output Files
For this example we will use the directory 02_assemblies previously created with the assemble module. We run the following Captus command to search and extract our reference markers from the assemblies:
captus extract -a 02_assemblies \
-n Angiosperms353 \
-p SeedPlantsPTD \
-m SeedPlantsMIT \
-d noncoding_DNA.fasta \
-c \
--max_loci_files 500
Warning
Notice the addition of option --max_loci_files 500, which is used only for showing the output directories containing separate FASTA file per marker (3’, 4’, 5’, 6’, 13’, and 14’ in the image below, not created by default), we don’t recommend using this option in your runs since it will unnecesarily create large numbers of small FASTA files which would have to be concatenated again anyways during the alignment step.
You can read more about the option here: –max_loci_files
After the run is finished we should see a new directory called 03_extractions with the following structure and files:
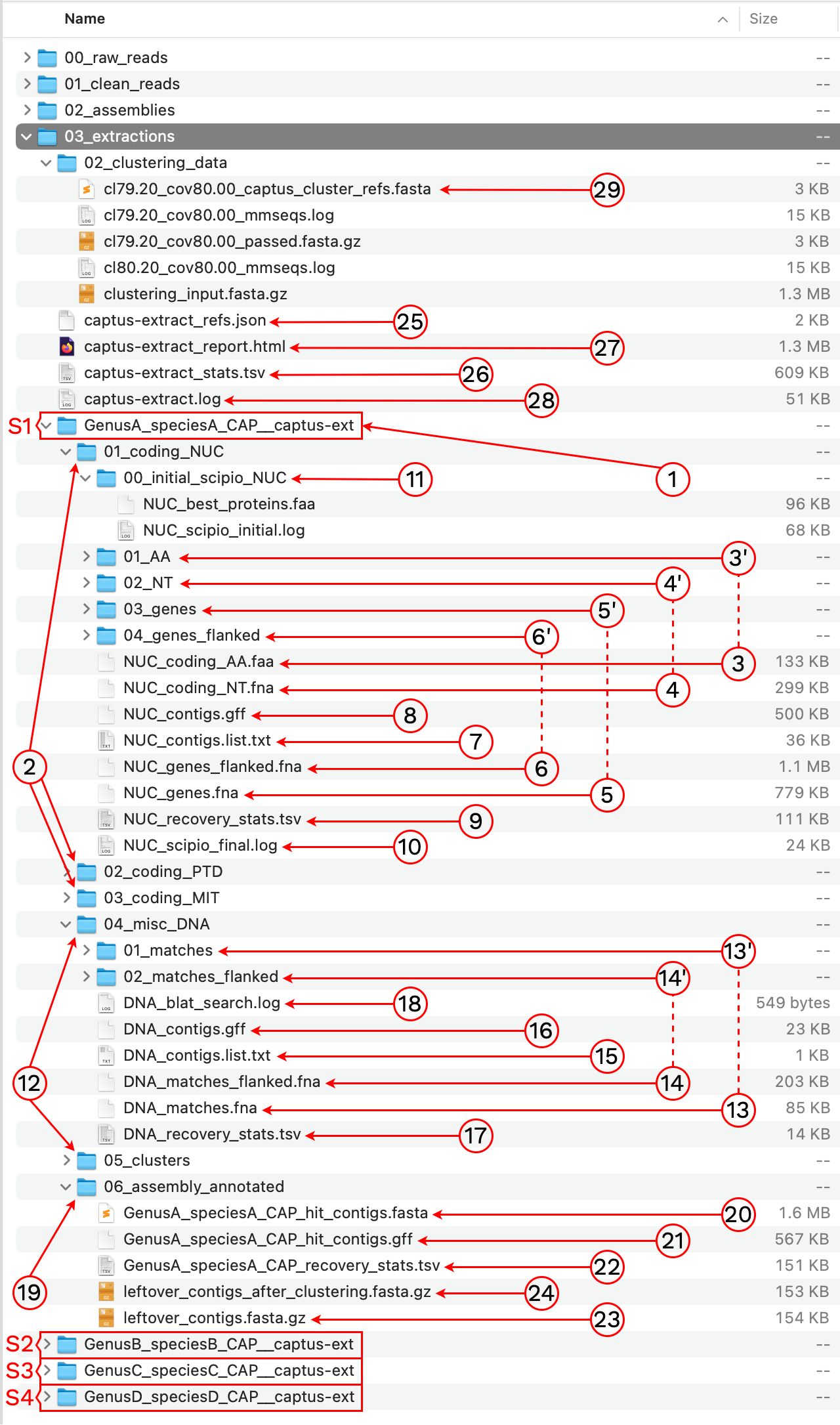
1. [SAMPLE_NAME]__captus-ext
A subdirectory ending in __captus-ext is created to contain the extracted markers of each sample separately (S1, S2, S3, and S4 in the image).
2. 01_coding_NUC, 02_coding_PTD, 03_coding_MIT
These directories contain the extracted coding markers from the NUClear, PlasTiDial, and MITochondrial genomes respectively.
The markers are presented in four formats: protein sequence (coding_AA), coding sequence in nucleotide (coding_NT), exons and introns concatenated (genes), and the concatenation of exons and introns flanked by a fixed length of sequence (genes_flanked):
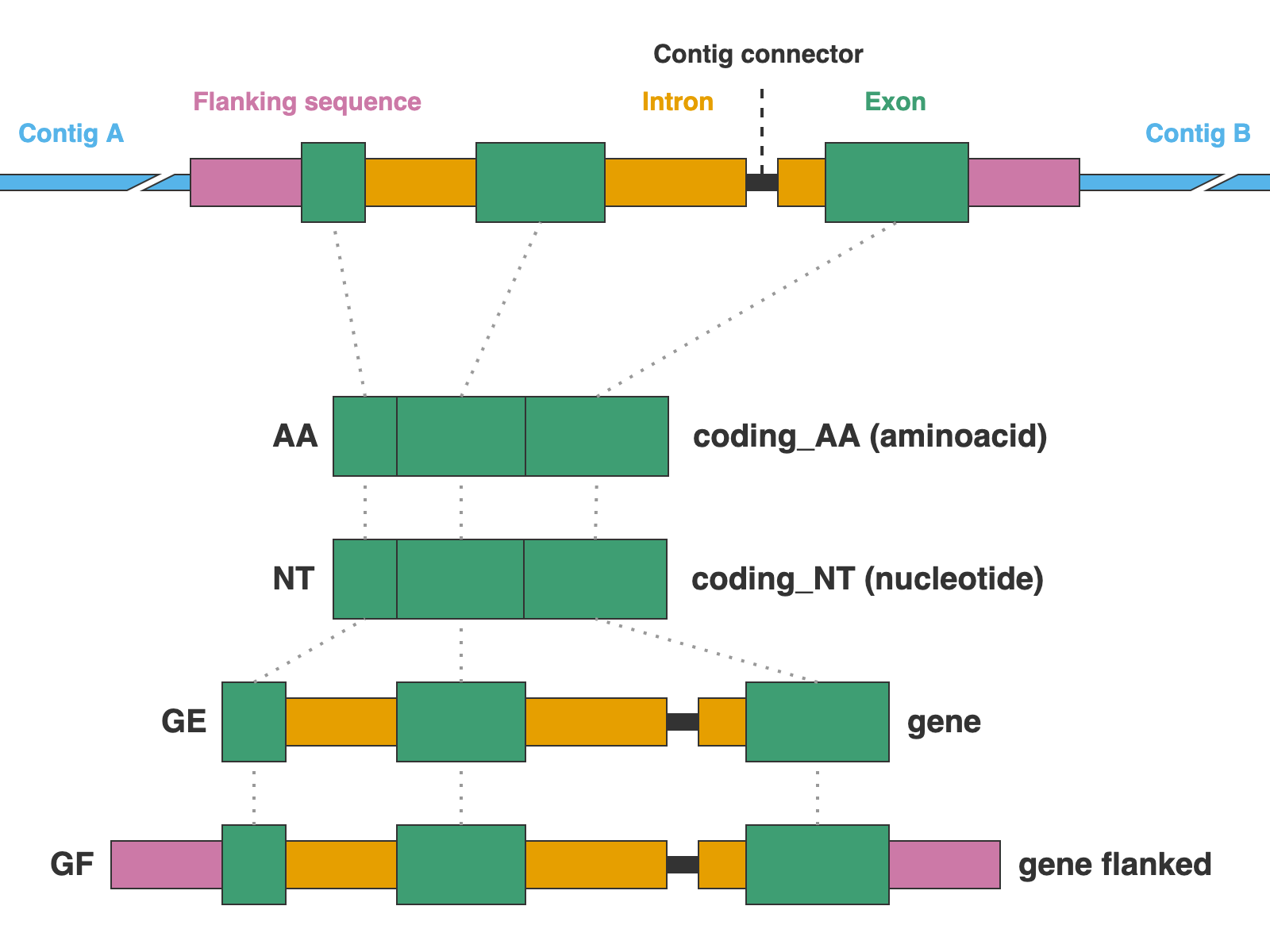
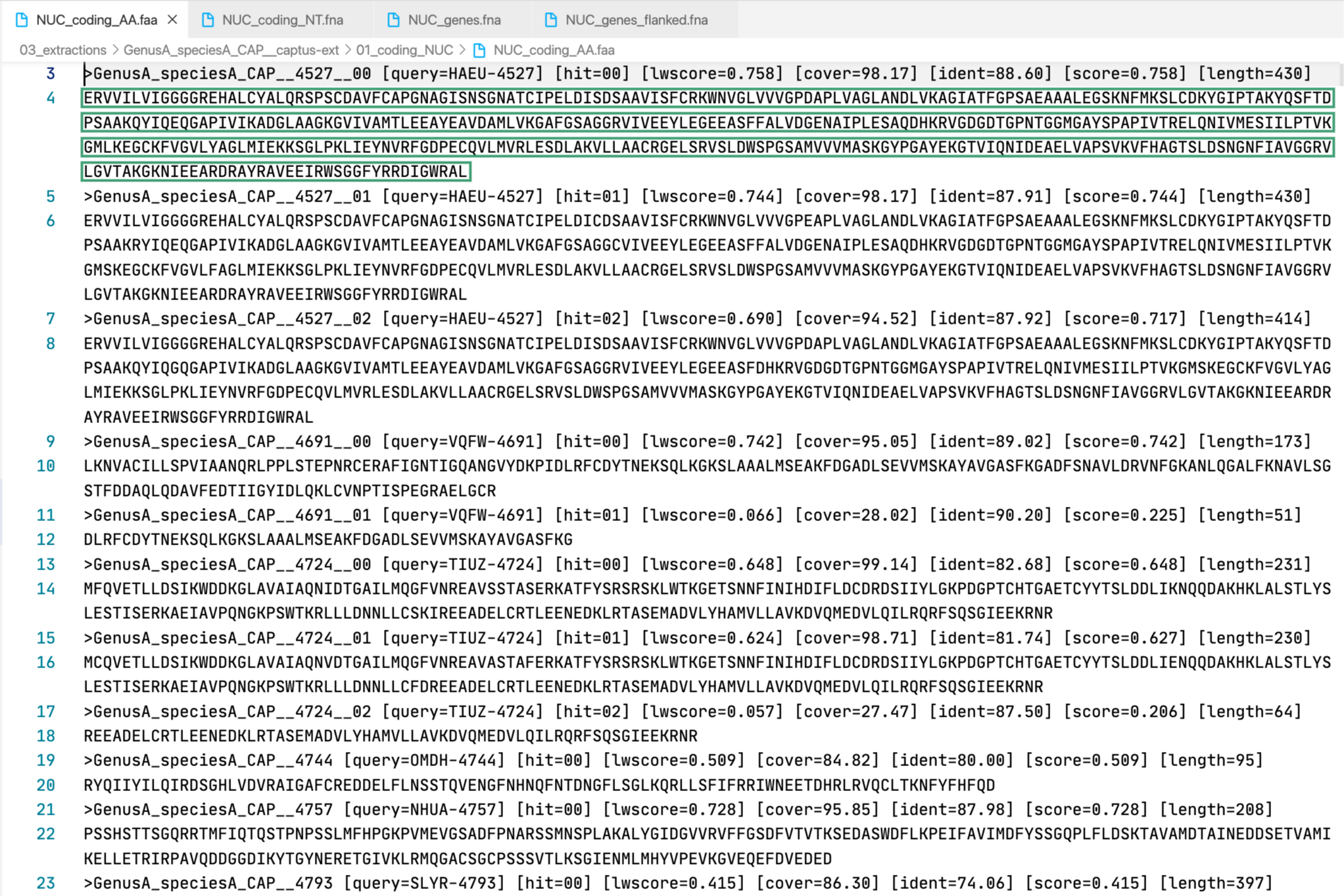
3. [MARKER_TYPE]_coding_AA.faa, 01_AA
Coding sequence in aminoacids. Prefixes can be NUC, PTD, or MIT. For details on sequence headers see FASTA headers explanation.
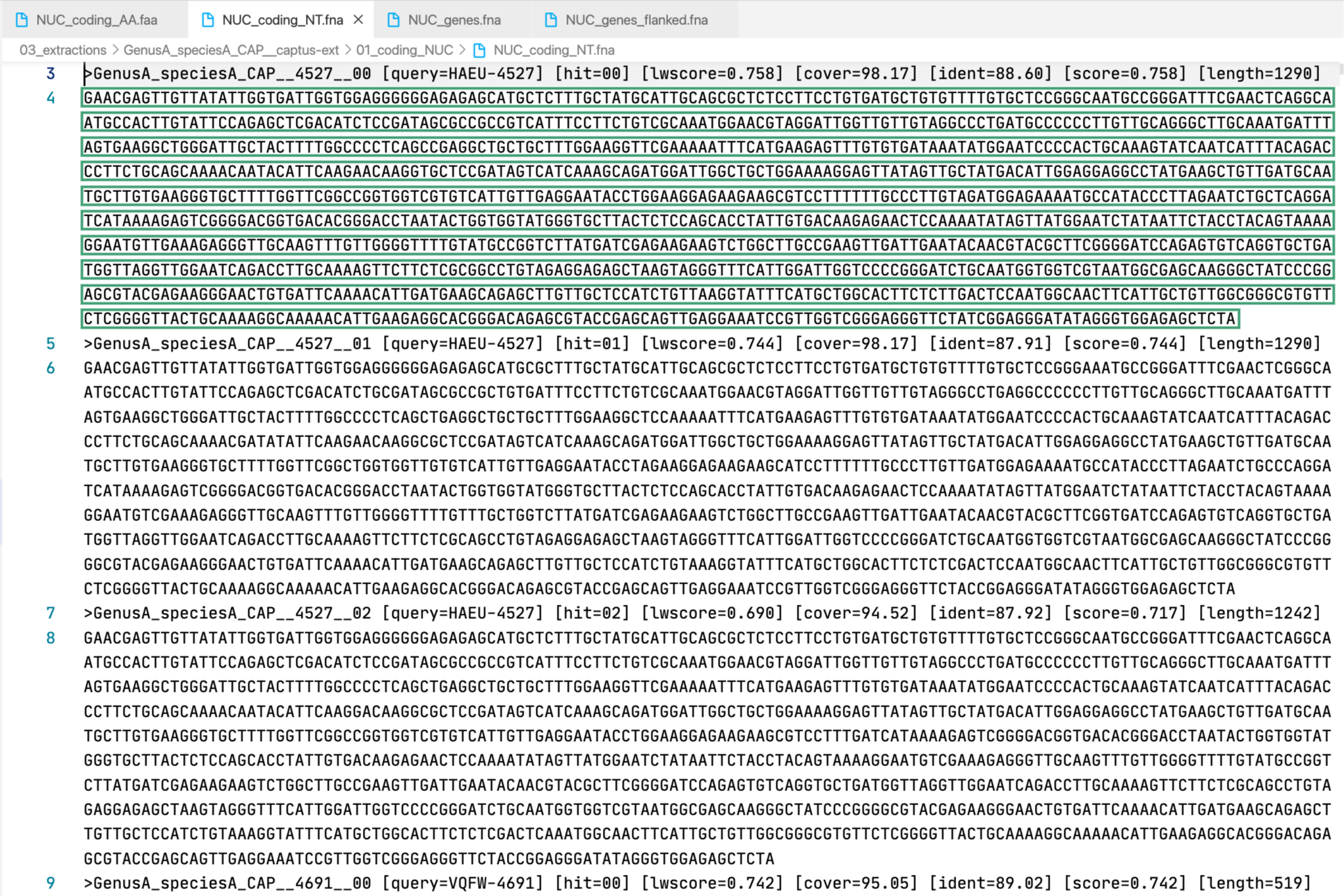
4. [MARKER_TYPE]_coding_NT.fna, 02_NT
Coding sequence in nucleotides, a.k.a. CDS. Prefixes can be NUC, PTD, or MIT. For details on sequence headers see FASTA headers explanation.
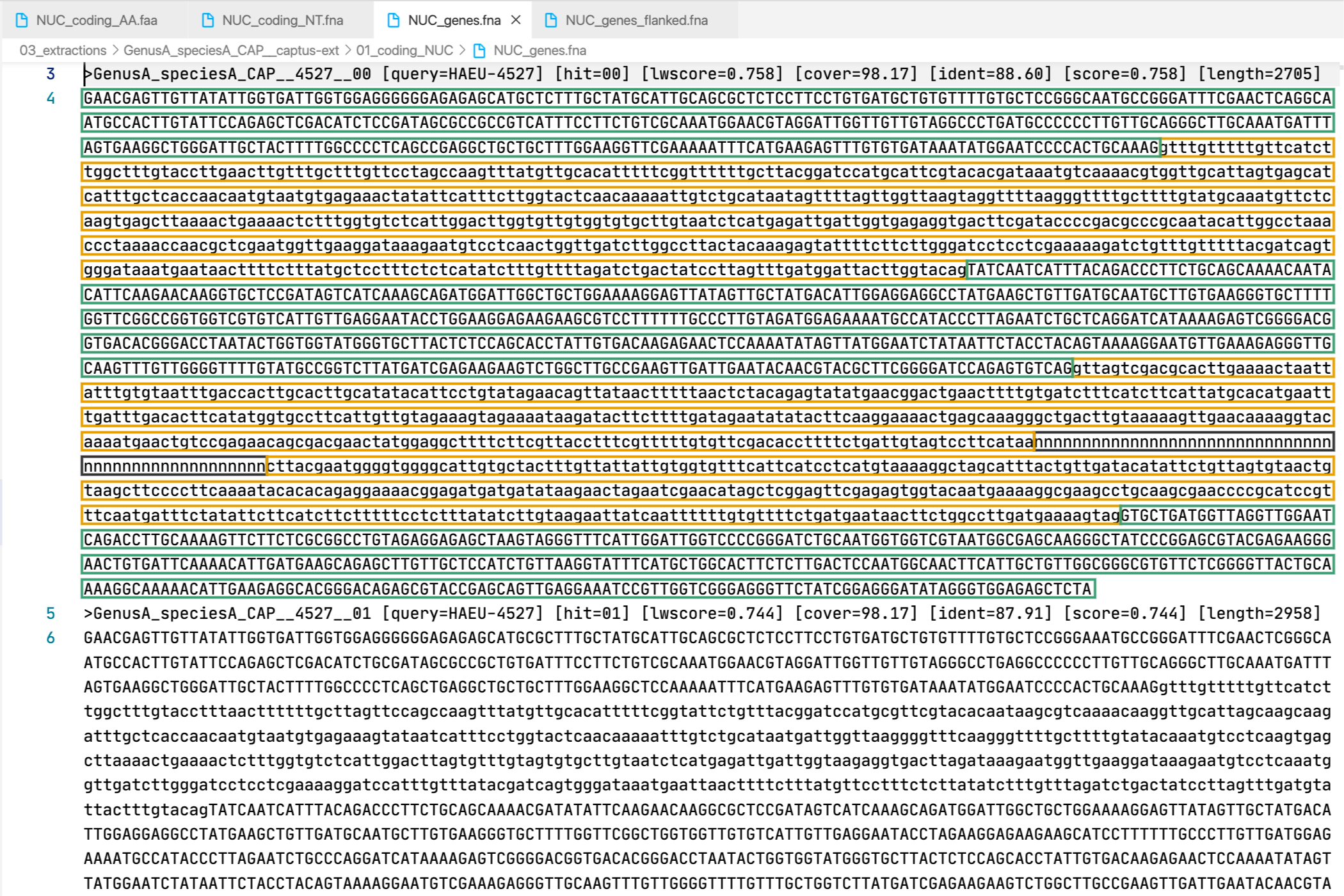
5. [MARKER_TYPE]_genes.fna, 03_genes
Gene sequence (exons in capital letters + introns in lowercase letters) in nucleotides. A contig connector of 50 n characters is included when the protein match spans more than a single contig. Prefixes can be NUC, PTD, or MIT. For details on sequence headers see FASTA headers explanation.
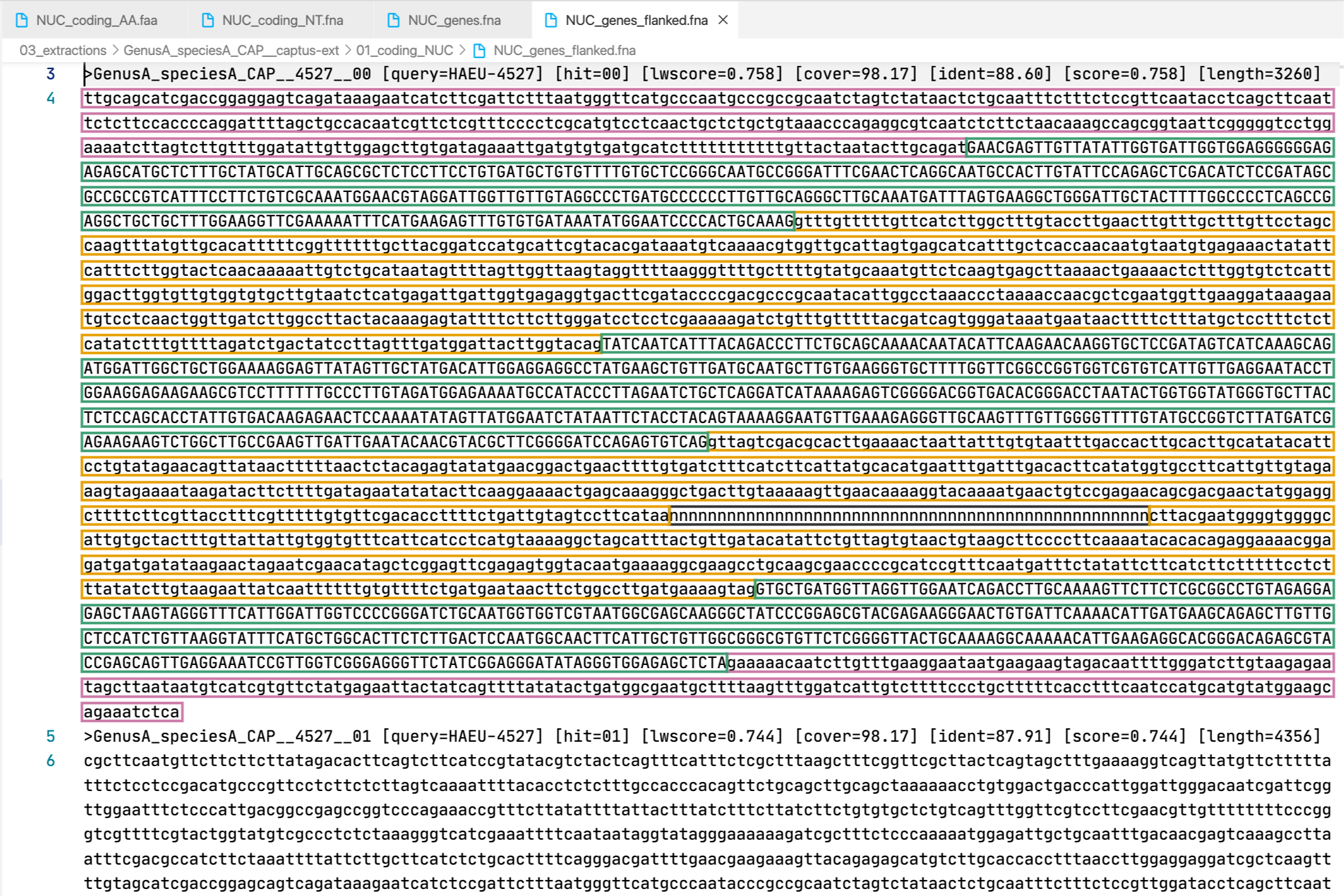
6. [MARKER_TYPE]_genes_flanked.fna, 04_genes_flanked
Gene sequence (exons in capital letters + introns in lowercase letters) plus additional flanking sequence in lowercase nucleotides. A contig connector of 50 n characters is included when the protein match spans more than a single contig. Prefixes can be NUC, PTD, or MIT. For details on sequence headers see FASTA headers explanation.
7. [MARKER_TYPE]_contigs_list.txt
List of contig names that had protein hits. Prefixes can be NUC, PTD, or MIT.
Example
NODE_138_length_18304_cov_18.0000_k_175_flag_1
NODE_635_length_5337_cov_17.0000_k_175_flag_1
NODE_46_length_16959_cov_19.0000_k_175_flag_1
NODE_321_length_4728_cov_10.0000_k_175_flag_1
NODE_760_length_19021_cov_17.9856_k_175_flag_0
NODE_621_length_11511_cov_11.9845_k_175_flag_0
NODE_965_length_6331_cov_15.0000_k_175_flag_1
NODE_948_length_26295_cov_58.0000_k_175_flag_0
NODE_1726_length_1438_cov_9.0000_k_175_flag_1
NODE_210_length_2896_cov_9.9471_k_175_flag_0
NODE_677_length_10733_cov_14.0000_k_175_flag_1
NODE_996_length_2375_cov_11.0000_k_175_flag_1
NODE_1837_length_366_cov_3.0000_k_175_flag_1
NODE_1647_length_529_cov_7.0000_k_175_flag_1
NODE_792_length_4384_cov_17.0000_k_175_flag_1
NODE_1378_length_5491_cov_21.0000_k_175_flag_1
NODE_602_length_14961_cov_47.0000_k_175_flag_1
NODE_1293_length_649_cov_3.0000_k_175_flag_1
NODE_949_length_2240_cov_50.0000_k_175_flag_1
NODE_751_length_2777_cov_34.7909_k_175_flag_0
8. [MARKER_TYPE]_contigs.gff
Annotation track in GFF format for protein hits to contigs in assembly. Prefixes can be NUC, PTD, or MIT.
See 19. 06_assembly_annotated
9. [MARKER_TYPE]_recovery_stats.tsv
Tab-separated-values table with marker recovery statistics, these are concatenated across marker types and samples and summarized in the final Marker Recovery report. Prefixes can be NUC, PTD, or MIT.
Information included in the table
| Column |
Description |
| sample_name |
Name of the sample. |
| marker_type |
Type of marker. Possible values are NUC, PTD, MIT, DNA, or CLR. |
| locus |
Name of the locus. |
| ref_name |
Name of the reference selected for the locus. Relevant when the reference contains multiple sequences per locus like in Angiosperms353 for example. |
| ref_coords |
Match coordinates with respect to the reference, each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ;. For example: 1-47;48-354,355-449 indicates that a contig contained a segment matching reference coordinates 1-49 and a different contig matched two segments, 48-354 and 355-449 respectively. |
| ref_type |
Whether the reference is an aminoacid (prot) or nucleotide (nucl) sequence. |
| ref_len_matched |
Number of residues matched in the reference. |
| hit |
Paralog ranking, 00 is assigned to the best hit, secondary hits start at 01. |
| pct_recovered |
Percentage of the total length of the reference sequence that was matched. |
| pct_identity |
Percentage of sequence identity between the hit and the reference sequence. |
| score |
Inspired by Scipio’s score: (matches - mismatches) / reference length. |
| wscore |
Weighted score. When the reference contains multiple sequences per locus, the best-matching reference is decided after normalizing their recovered length across references in the locus and multiplying that value by their respective score, thus producing the wscore. Finally wscore is also penalized by the number of frameshifts (if the marker is coding) and number of contigs used in the assembly of the hit. |
| hit_len |
Number of residues matched in the sample’s contig(s) plus the length of the flanking sequence. |
| cds_len |
If ref_type is prot this number represents the number of residues corresponding to coding sequence (i.e. exons). If the ref_type is nucl this field shows NA. |
| intron_len |
If ref_type is prot this number represents the number of residues corresponding to intervening non-coding sequence segments (i.e. introns). If the ref_type is nucl this field shows NA. |
| flanks_len |
Number of residues included in the flanking sequence. |
| frameshifts |
Positions of the corrected frameshifts in the output sequence. If the ref_type is nucl this field shows NA. |
| hit_contigs |
Number of contigs used to assemble the hit. |
| hit_l50 |
Least number of contigs in the hit that contain 50% of the recovered length. |
| hit_l90 |
Least number of contigs in the hit that contain 90% of the recovered length. |
| hit_lg50 |
Least number of contigs in the hit that contain 50% of the reference locus length. |
| hit_lg90 |
Least number of contigs in the hit that contain 90% of the reference locus length. |
| ctg_names |
Name of the contigs used in the reconstruction of the hit. Example: NODE_6256_length_619_cov_3.0000_k_169_flag_1;NODE_3991_length_1778_cov_19.0000_k_169_flag_1, for a hit where two contigs were used. |
| ctg_strands |
Contig strands (+ or -) provided in the same order as ctg_names. Example: +;- indicates that the contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 was matched in the positive strand while the contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 was matched in the ngeative strand. |
| ctg_coords |
Match coordinates with respect to the contigs in the sample’s assembly. Each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ; which are provided in the same order as ctg_names and ctg_strands. Example: 303-452;694-1626,301-597 indicates that a single segment was matched in contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 in the + strand with coordinates 303-452, while two segments were matched in contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 in the - strand with coordinates 694-1626 and 301-597 respectively. |
10. [MARKER_TYPE]_scipio_final.log
Log of the second Scipio’s run, where best references have already been selected (when using multi-sequence per locus references) and only the contigs that had hits durin Scipio’s initial run are used. Prefixes can be NUC, PTD, or MIT.
11. 00_initial_scipio_[MARKER_TYPE]
Directory for Scipio’s initial run results. The directory contains the set of filtered protein references [MARKER_TYPE]_best_proteins.faa (when using multi-sequence per locus references) and the log of Scipio’s initial run [MARKER_TYPE]_scipio_initial.log. Suffixes can be NUC, PTD, or MIT.
12. 04_misc_DNA, 05_clusters
These directories contain the extracted miscellaneous DNA markers, either from a DNA custom set of references or from the CLusteRing resulting from using the option --cluster_leftovers.
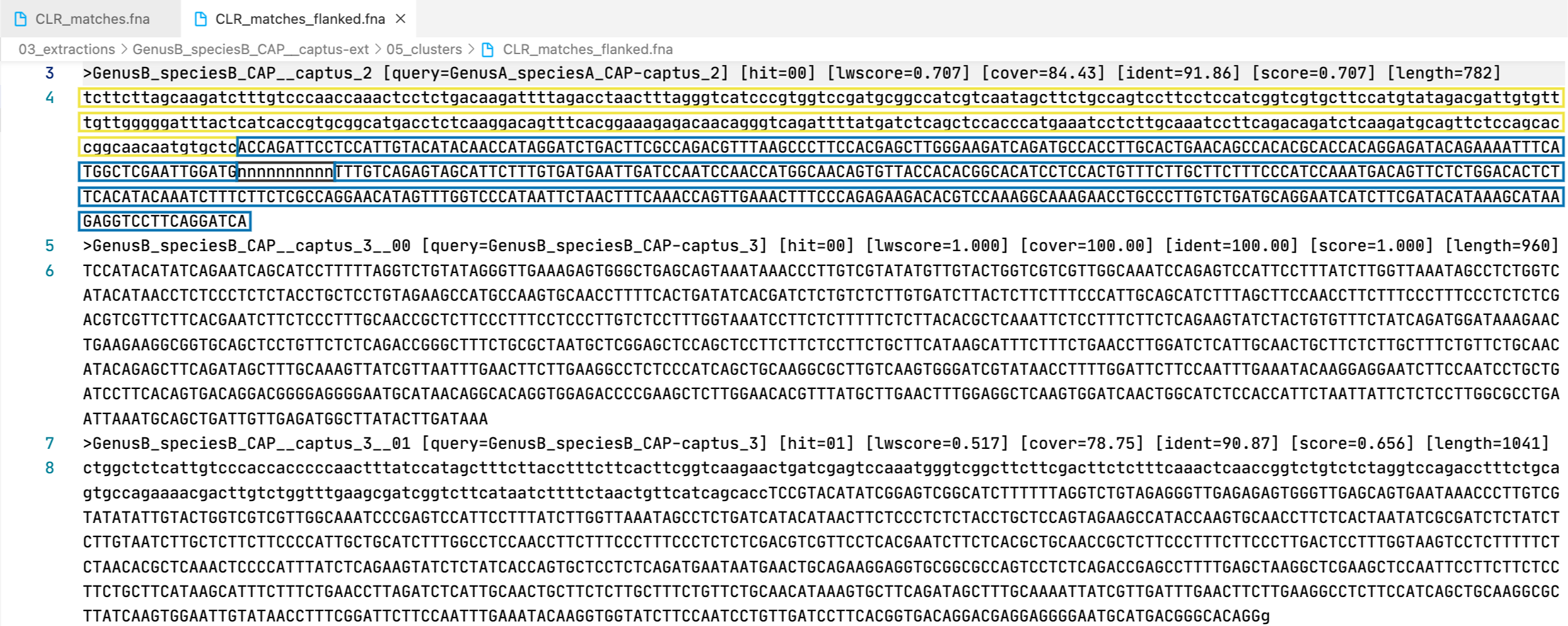
The markers are presented in two formats: matching DNA segments (matches), and the matched segments including flanks and other intervening segments not present in the reference (matches_flanked).
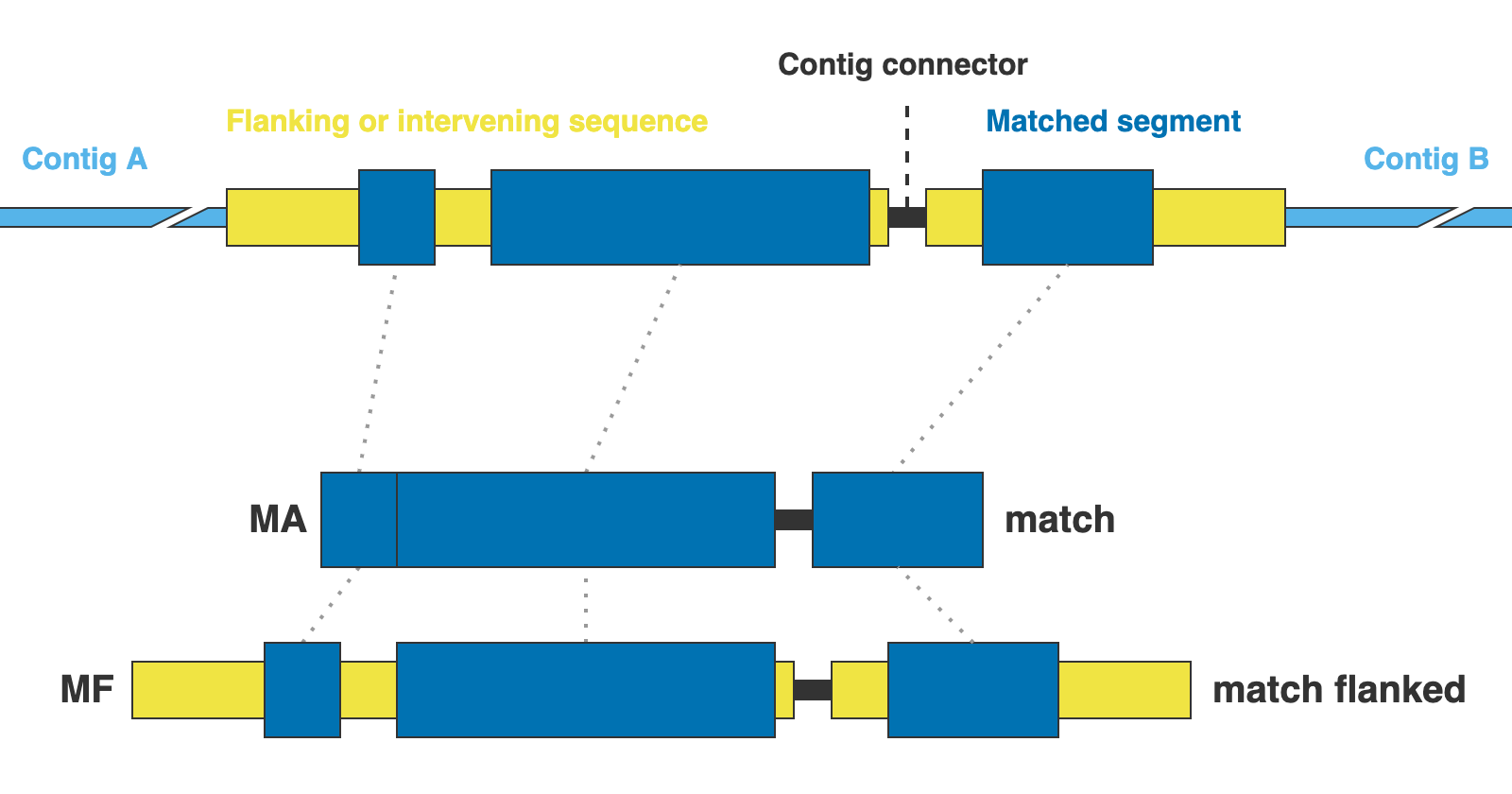
13. [MARKER_TYPE]_matches.fna, 01_matches
Matches per miscellaneous DNA marker in nucleotides. Prefixes can be DNA or CLR. For details on sequence headers see FASTA headers explanation.
14. [MARKER_TYPE]_matches_flanked.fna, 02_matches_flanked
Matches plus additional flanking sequence per miscellaneous DNA marker in nucleotides. Prefixes can be DNA or CLR. For details on sequence headers see FASTA headers explanation.
15. [MARKER_TYPE]_contigs_list.txt
List of contig names that had miscellaneous DNA marker hits. Prefixes can be DNA or CLR.
Example
NODE_1858_length_3636_cov_14.0000_k_175_flag_1
NODE_2876_length_3179_cov_25.0000_k_175_flag_1
NODE_502_length_5771_cov_37.0000_k_175_flag_1
NODE_347_length_475_cov_2.0000_k_175_flag_1
NODE_1393_length_297_cov_4.0000_k_175_flag_1
NODE_3093_length_960_cov_17.0000_k_175_flag_1
NODE_3265_length_1041_cov_18.0000_k_175_flag_1
16. [MARKER_TYPE]_contigs.gff
Annotation track in GFF format for miscellaneous DNA marker hits to contigs in assembly. Prefixes can be DNA or CLR.
See 19. 06_assembly_annotated
17. [MARKER_TYPE]_recovery_stats.tsv
Tab-separated-values table with marker recovery statistics, these are concatenated across marker types and samples and summarized in the final Marker Recovery report. Prefixes can be DNA or CLR.
Information included in the table
| Column |
Description |
| sample_name |
Name of the sample. |
| marker_type |
Type of marker. Possible values are NUC, PTD, MIT, DNA, or CLR. |
| locus |
Name of the locus. |
| ref_name |
Name of the reference selected for the locus. Relevant when the reference contains multiple sequences per locus like in Angiosperms353 for example. |
| ref_coords |
Match coordinates with respect to the reference, each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ;. For example: 1-47;48-354,355-449 indicates that a contig contained a segment matching reference coordinates 1-49 and a different contig matched two segments, 48-354 and 355-449 respectively. |
| ref_type |
Whether the reference is an aminoacid (prot) or nucleotide (nucl) sequence. |
| ref_len_matched |
Number of residues matched in the reference. |
| hit |
Paralog ranking, 00 is assigned to the best hit, secondary hits start at 01. |
| pct_recovered |
Percentage of the total length of the reference sequence that was matched. |
| pct_identity |
Percentage of sequence identity between the hit and the reference sequence. |
| score |
Inspired by Scipio’s score: (matches - mismatches) / reference length. |
| wscore |
Weighted score. When the reference contains multiple sequences per locus, the best-matching reference is decided after normalizing their recovered length across references in the locus and multiplying that value by their respective score, thus producing the wscore. Finally wscore is also penalized by the number of frameshifts (if the marker is coding) and number of contigs used in the assembly of the hit. |
| hit_len |
Number of residues matched in the sample’s contig(s) plus the length of the flanking sequence. |
| cds_len |
If ref_type is prot this number represents the number of residues corresponding to coding sequence (i.e. exons). If the ref_type is nucl this field shows NA. |
| intron_len |
If ref_type is prot this number represents the number of residues corresponding to intervening non-coding sequence segments (i.e. introns). If the ref_type is nucl this field shows NA. |
| flanks_len |
Number of residues included in the flanking sequence. |
| frameshifts |
Positions of the corrected frameshifts in the output sequence. If the ref_type is nucl this field shows NA. |
| hit_contigs |
Number of contigs used to assemble the hit. |
| hit_l50 |
Least number of contigs in the hit that contain 50% of the recovered length. |
| hit_l90 |
Least number of contigs in the hit that contain 90% of the recovered length. |
| hit_lg50 |
Least number of contigs in the hit that contain 50% of the reference locus length. |
| hit_lg90 |
Least number of contigs in the hit that contain 90% of the reference locus length. |
| ctg_names |
Name of the contigs used in the reconstruction of the hit. Example: NODE_6256_length_619_cov_3.0000_k_169_flag_1;NODE_3991_length_1778_cov_19.0000_k_169_flag_1, for a hit where two contigs were used. |
| ctg_strands |
Contig strands (+ or -) provided in the same order as ctg_names. Example: +;- indicates that the contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 was matched in the positive strand while the contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 was matched in the ngeative strand. |
| ctg_coords |
Match coordinates with respect to the contigs in the sample’s assembly. Each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ; which are provided in the same order as ctg_names and ctg_strands. Example: 303-452;694-1626,301-597 indicates that a single segment was matched in contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 in the + strand with coordinates 303-452, while two segments were matched in contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 in the - strand with coordinates 694-1626 and 301-597 respectively. |
18. [MARKER_TYPE]_blat_search.log
Log of BLAT’s run. Prefixes can be DNA or CLR.
19. 06_assembly_annotated
The main outputs of this directory are a FASTA file containing all the contigs that had hits to the reference markers called [SAMPLE_NAME]_hit_contigs.fasta as well as an annotation track for those markers called [SAMPLE_NAME]_hit_contigs.gff. You can visualize the annotations in Geneious, for example, by importing the FASTA file and then dropping the GFF file on top:
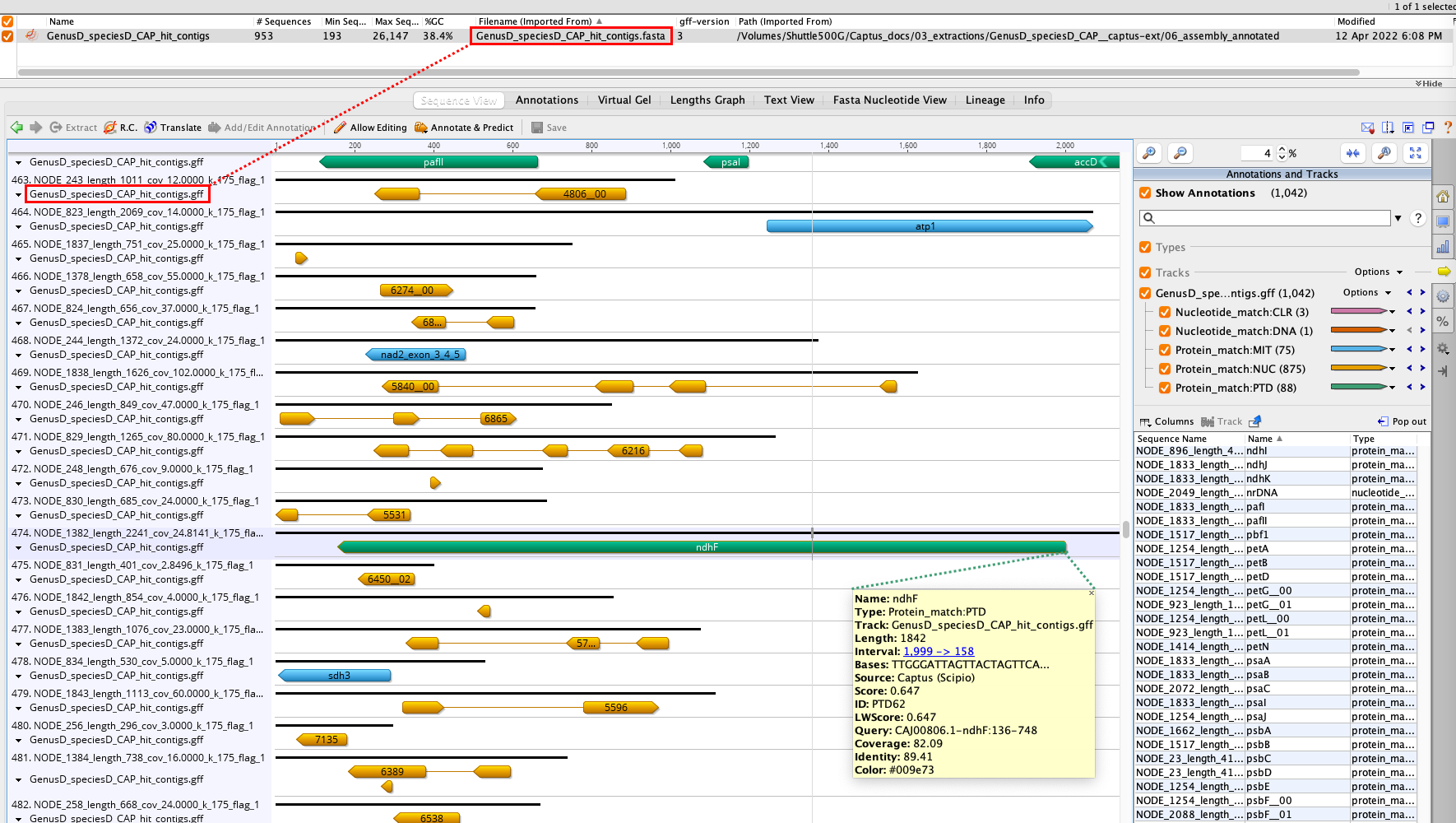
20. [SAMPLE_NAME]_hit_contigs.fasta
This file contains the subset of the contigs assembled by MEGAHIT that had hit to the reference markers. See the red rectangles in 19. 06_assembly_annotated.
21. [SAMPLE_NAME]_hit_contigs.gff
Unified annotation track in GFF format for ALL the marker types found in the assembly’s contigs. See the red rectangles in 19. 06_assembly_annotated.
22. [SAMPLE_NAME]_recovery_stats.tsv
Unified tab-separated-values table with marker recovery statistics from ALL the marker types found in the sample, these are concatenated across samples and summarized in the final Marker Recovery report.
Information included in the table
| Column |
Description |
| sample_name |
Name of the sample. |
| marker_type |
Type of marker. Possible values are NUC, PTD, MIT, DNA, or CLR. |
| locus |
Name of the locus. |
| ref_name |
Name of the reference selected for the locus. Relevant when the reference contains multiple sequences per locus like in Angiosperms353 for example. |
| ref_coords |
Match coordinates with respect to the reference, each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ;. For example: 1-47;48-354,355-449 indicates that a contig contained a segment matching reference coordinates 1-49 and a different contig matched two segments, 48-354 and 355-449 respectively. |
| ref_type |
Whether the reference is an aminoacid (prot) or nucleotide (nucl) sequence. |
| ref_len_matched |
Number of residues matched in the reference. |
| hit |
Paralog ranking, 00 is assigned to the best hit, secondary hits start at 01. |
| pct_recovered |
Percentage of the total length of the reference sequence that was matched. |
| pct_identity |
Percentage of sequence identity between the hit and the reference sequence. |
| score |
Inspired by Scipio’s score: (matches - mismatches) / reference length. |
| wscore |
Weighted score. When the reference contains multiple sequences per locus, the best-matching reference is decided after normalizing their recovered length across references in the locus and multiplying that value by their respective score, thus producing the wscore. Finally wscore is also penalized by the number of frameshifts (if the marker is coding) and number of contigs used in the assembly of the hit. |
| hit_len |
Number of residues matched in the sample’s contig(s) plus the length of the flanking sequence. |
| cds_len |
If ref_type is prot this number represents the number of residues corresponding to coding sequence (i.e. exons). If the ref_type is nucl this field shows NA. |
| intron_len |
If ref_type is prot this number represents the number of residues corresponding to intervening non-coding sequence segments (i.e. introns). If the ref_type is nucl this field shows NA. |
| flanks_len |
Number of residues included in the flanking sequence. |
| frameshifts |
Positions of the corrected frameshifts in the output sequence. If the ref_type is nucl this field shows NA. |
| hit_contigs |
Number of contigs used to assemble the hit. |
| hit_l50 |
Least number of contigs in the hit that contain 50% of the recovered length. |
| hit_l90 |
Least number of contigs in the hit that contain 90% of the recovered length. |
| hit_lg50 |
Least number of contigs in the hit that contain 50% of the reference locus length. |
| hit_lg90 |
Least number of contigs in the hit that contain 90% of the reference locus length. |
| ctg_names |
Name of the contigs used in the reconstruction of the hit. Example: NODE_6256_length_619_cov_3.0000_k_169_flag_1;NODE_3991_length_1778_cov_19.0000_k_169_flag_1, for a hit where two contigs were used. |
| ctg_strands |
Contig strands (+ or -) provided in the same order as ctg_names. Example: +;- indicates that the contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 was matched in the positive strand while the contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 was matched in the ngeative strand. |
| ctg_coords |
Match coordinates with respect to the contigs in the sample’s assembly. Each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ; which are provided in the same order as ctg_names and ctg_strands. Example: 303-452;694-1626,301-597 indicates that a single segment was matched in contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 in the + strand with coordinates 303-452, while two segments were matched in contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 in the - strand with coordinates 694-1626 and 301-597 respectively. |
23. leftover_contigs.fasta.gz
This file contains the subset of the contigs assembled by MEGAHIT that had no hit to the reference markers. The file is compressed to save space. These are the contigs that are used for clustering across samples in order to discover additional homologous markers.
24. leftover_contigs_after_custering.fasta.gz
This file contains the subset of the contigs assembled by MEGAHIT that had no hit to the reference markers or even to the newly discovered markers derived from clusterin. The file is compressed to save space.
This file stores the paths to all the references used for extraction. This file is necessary so the alignment step can correctly add the references to the final alignments to be used as guides.
Example
{
"NUC": {
"AA_path": "/Users/emortiz/software/GitHub/Captus/data/Angiosperms353.FAA",
"AA_msg": "Angiosperms353 /Users/emortiz/software/GitHub/Captus/data/Angiosperms353.FAA",
"NT_path": "/Users/emortiz/software/GitHub/Captus/data/Angiosperms353.FNA",
"NT_msg": "Angiosperms353 /Users/emortiz/software/GitHub/Captus/data/Angiosperms353.FNA"
},
"PTD": {
"AA_path": "/Users/emortiz/software/GitHub/Captus/data/SeedPlantsPTD.FAA",
"AA_msg": "SeedPlantsPTD /Users/emortiz/software/GitHub/Captus/data/SeedPlantsPTD.FAA",
"NT_path": "/Users/emortiz/software/GitHub/Captus/data/SeedPlantsPTD.FNA",
"NT_msg": "SeedPlantsPTD /Users/emortiz/software/GitHub/Captus/data/SeedPlantsPTD.FNA"
},
"MIT": {
"AA_path": "/Users/emortiz/software/GitHub/Captus/data/SeedPlantsMIT.FAA",
"AA_msg": "SeedPlantsMIT /Users/emortiz/software/GitHub/Captus/data/SeedPlantsMIT.FAA",
"NT_path": "/Users/emortiz/software/GitHub/Captus/data/SeedPlantsMIT.FNA",
"NT_msg": "SeedPlantsMIT /Users/emortiz/software/GitHub/Captus/data/SeedPlantsMIT.FNA"
},
"DNA": {
"AA_path": null,
"AA_msg": null,
"NT_path": "/Volumes/Shuttle500G/for_docs_output/nrDNA.fasta",
"NT_msg": "/Volumes/Shuttle500G/for_docs_output/nrDNA.fasta"
},
"CLR": {
"AA_path": null,
"AA_msg": null,
"NT_path": "/Volumes/Shuttle500G/for_docs_output/03_extractions/01_clustering_data/clust_id79.20_cov80.00_captus_clusters_refs.fasta",
"NT_msg": "/Volumes/Shuttle500G/for_docs_output/03_extractions/01_clustering_data/clust_id79.20_cov80.00_captus_clusters_refs.fasta"
}
}
Unified tab-separated-values table with marker recovery statistics from ALL the markers found in ALL the samples, this table is used to create the final Marker Recovery report. Even though the report is quite useful for visualization you might need to do more complex statistical analysis, this table is the most appropriate output file for such analyses.
Information included in the table
| Column |
Description |
| sample_name |
Name of the sample. |
| marker_type |
Type of marker. Possible values are NUC, PTD, MIT, DNA, or CLR. |
| locus |
Name of the locus. |
| ref_name |
Name of the reference selected for the locus. Relevant when the reference contains multiple sequences per locus like in Angiosperms353 for example. |
| ref_coords |
Match coordinates with respect to the reference, each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ;. For example: 1-47;48-354,355-449 indicates that a contig contained a segment matching reference coordinates 1-49 and a different contig matched two segments, 48-354 and 355-449 respectively. |
| ref_type |
Whether the reference is an aminoacid (prot) or nucleotide (nucl) sequence. |
| ref_len_matched |
Number of residues matched in the reference. |
| hit |
Paralog ranking, 00 is assigned to the best hit, secondary hits start at 01. |
| pct_recovered |
Percentage of the total length of the reference sequence that was matched. |
| pct_identity |
Percentage of sequence identity between the hit and the reference sequence. |
| score |
Inspired by Scipio’s score: (matches - mismatches) / reference length. |
| wscore |
Weighted score. When the reference contains multiple sequences per locus, the best-matching reference is decided after normalizing their recovered length across references in the locus and multiplying that value by their respective score, thus producing the wscore. Finally wscore is also penalized by the number of frameshifts (if the marker is coding) and number of contigs used in the assembly of the hit. |
| hit_len |
Number of residues matched in the sample’s contig(s) plus the length of the flanking sequence. |
| cds_len |
If ref_type is prot this number represents the number of residues corresponding to coding sequence (i.e. exons). If the ref_type is nucl this field shows NA. |
| intron_len |
If ref_type is prot this number represents the number of residues corresponding to intervening non-coding sequence segments (i.e. introns). If the ref_type is nucl this field shows NA. |
| flanks_len |
Number of residues included in the flanking sequence. |
| frameshifts |
Positions of the corrected frameshifts in the output sequence. If the ref_type is nucl this field shows NA. |
| hit_contigs |
Number of contigs used to assemble the hit. |
| hit_l50 |
Least number of contigs in the hit that contain 50% of the recovered length. |
| hit_l90 |
Least number of contigs in the hit that contain 90% of the recovered length. |
| hit_lg50 |
Least number of contigs in the hit that contain 50% of the reference locus length. |
| hit_lg90 |
Least number of contigs in the hit that contain 90% of the reference locus length. |
| ctg_names |
Name of the contigs used in the reconstruction of the hit. Example: NODE_6256_length_619_cov_3.0000_k_169_flag_1;NODE_3991_length_1778_cov_19.0000_k_169_flag_1, for a hit where two contigs were used. |
| ctg_strands |
Contig strands (+ or -) provided in the same order as ctg_names. Example: +;- indicates that the contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 was matched in the positive strand while the contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 was matched in the ngeative strand. |
| ctg_coords |
Match coordinates with respect to the contigs in the sample’s assembly. Each segment is expressed as [start]-[end], segments within the same contig are separated by ,, and segments in different contigs are separated by ; which are provided in the same order as ctg_names and ctg_strands. Example: 303-452;694-1626,301-597 indicates that a single segment was matched in contig NODE_6256_length_619_cov_3.0000_k_169_flag_1 in the + strand with coordinates 303-452, while two segments were matched in contig NODE_3991_length_1778_cov_19.0000_k_169_flag_1 in the - strand with coordinates 694-1626 and 301-597 respectively. |
This is the final Marker Recovery report, summarizing marker extraction statistics across all samples and marker types.
This is the log from Captus, it contains the command used and all the information shown during the run. If the option --show_less was enabled, the log will also contain all the extra detailed information that was hidden during the run.
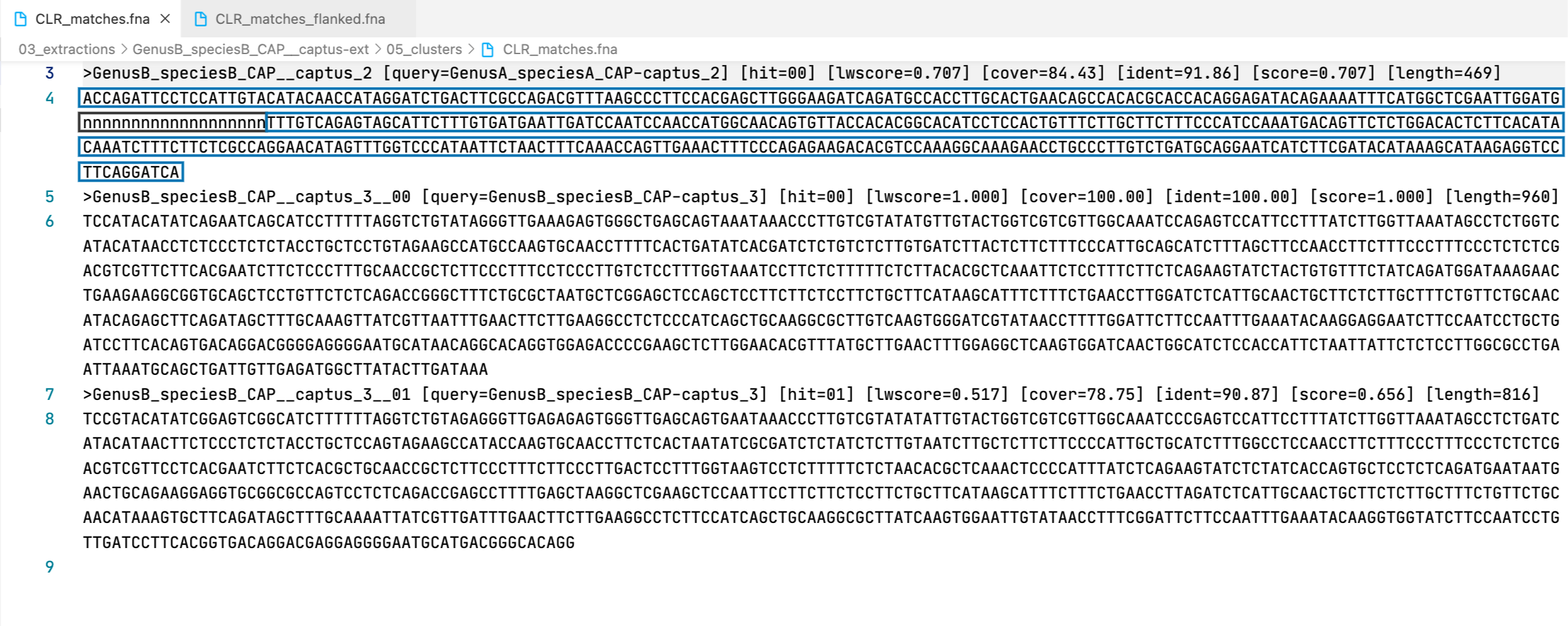
29. clust_id##.##_cov##.##_captus_clusters_refs.fasta
This FASTA file contains the cluster representatives that will be searched and extracted across samples (prefix CLR), the loci names are called captus_#. These represent newly discovered homologus markers in contigs that had no hits to other reference proteins or miscellaneous DNA markers.
Example
>GenusA_speciesA_CAP-captus_1
GACTTGAGCCCCAAAACTAGGTTGGGTGCAGGGGGTCGATCTTGATTTTATTACTCAGGGTGCTTCAGATCAGGTTCTTGCAGCTGAACATGCTTCGGGACATCGACCCTATGGTCAGAATCTTCAATCTGGAAGCTCTGCTGGTGCATCAAGCCAACAAGACATGTCCAAGATCATATGCCAGTAAGAAAACAAGGTATGTAAATACCTCTGCGGGTGGTGCTTTACCTGGAACAGCGTCTGAATCGACTGGCGATATCCCTGCTACTGATGATACCCCCCTGATGGATGCTGCAGGGTAAACAGGTTAATATAGATCTGCTATCTCGAGCCCTGTAACCTGGCTATGCTCTCTTTGCGTCTGCTACTAGATATGCTTCTGGCTTTGCCATTTCTATGAATTCTTATAGAATTTTGATTCAGTCACTATGCAAATTTTATTTGTTATGTTTATAAGGTCTCTTGATTGCGCTCGGGCTCCTGTCTCGGGGGAGCCCTGCGCTCCCTACGCTTACTAGAATATTCATTCTCCCCTATTAAAATAAAAATTTATCGAGATAAAAAAAGAA
>GenusA_speciesA_CAP-captus_2
ACCAGATTCCTCCATTGTACAGACAACCATAGGGTCCGACTTTGCAAGACGTTTAAGGCCTTCCACAAGCTTGGGAAGGTCAAATGCCACCTTGCACTGGACAGCCACACGTACCACAGGAGATACAGAAAATTTCATGGCTCGAATTGGATGGGCATCAACTTCCTTCTCGTTTGTTAGAGTGGCATTCTTGGTGATAAATTGGTCCAACCCAACCATGGCAACAGTATTACCACAAGGCACATCCTCCACCGTCTCTTGCCTCTTCCCCATCCAAATAACAGTTCTCTGGACACTCTTCACATACAGGTCTTTCTTCTCTCCAGGAACATAGTTTGGACCCATGATTCTAACTTTCAAACCGGTTGAAACTTTCCCAGAAAAGACTCGACCAAAGGCAAAGAACCTGCCCTTGTCTGATGCAGGAATCATCTTTGAGACATAGAGCATAAGAGGTCCCTCAGGGTCACAATTTCTAATGGCACTAGCATATGCGTCATCTAGTGGACCCTCATACAAATTCTCAACACGAT
>GenusB_speciesB_CAP-captus_3
TCCATACATATCAGAATCAGCATCCTTTTTAGGTCTGTATAGGGTTGAAAGAGTGGGCTGAGCAGTAAATAAACCCTTGTCGTATATGTTGTACTGGTCGTCGTTGGCAAATCCAGAGTCCATTCCTTTATCTTGGTTAAATAGCCTCTGGTCATACATAACCTCTCCCTCTCTACCTGCTCCTGTAGAAGCCATGCCAAGTGCAACCTTTTCACTGATATCACGATCTCTGTCTCTTGTGATCTTACTCTTCTTTCCCATTGCAGCATCTTTAGCTTCCAACCTTCTTTCCCTTTCCCTCTCTCGACGTCGTTCTTCACGAATCTTCTCCCTTTGCAACCGCTCTTCCCTTTCCTCCCTTGTCTCCTTTGGTAAATCCTTCTCTTTTTCTCTTACACGCTCAAATTCTCCTTTCTTCTCAGAAGTATCTACTGTGTTTCTATCAGATGGATAAAGAACTGAAGAAGGCGGTGCAGCTCCTGTTCTCTCAGACCGGGCTTTCTGCGCTAATGCTCGGAGCTCCAGCTCCTTCTTCTCCTTCTGCTTCATAAGCATTTCTTTCTGAACCTTGGATCTCATTGCAACTGCTTCTCTTGCTTTCTGTTCTGCAACATACAGAGCTTCAGATAGCTTTGCAAAGTTATCGTTAATTTGAACTTCTTGAAGGCCTCTCCCATCAGCTGCAAGGCGCTTGTCAAGTGGGATCGTATAACCTTTTGGATTCTTCCAATTTGAAATACAAGGAGGAATCTTCCAATCCTGCTGATCCTTCACAGTGACAGGACGGGGAGGGGAATGCATAACAGGCACAGGTGGAGACCCCGAAGCTCTTGGAACACGTTTATGCTTGAACTTTGGAGGCTCAAGTGGATCAACTGGCATCTCCACCATTCTAATTATTCTCTCCTTGGCGCCTGAATTAAATGCAGCTGATTGTTGAGATGGCTTATACTTGATAAA
Info
All the FASTA files produced by Captus containing extracted markers follow the same header style:
 Paralogs are ranked according to their
Paralogs are ranked according to their wscore which, in turn, is calculated from the percent identity (ident) as well as the percent coverage (cover) with respect to the selected reference sequence (query). The best hit is always ranked 00 and secondary hits start at 01. When a single hit is found for a marker (like in locus 5859 in the image) the ranking 00 is not included in the sequence name, only when multiple hits are found for a marker (like in locus 5865 in the image) the ranking 00 or 01 is included in the sequence name to make them unique. The description field frameshifts is only present when the output sequence has corrected frameshifts and the numbers indicate their position in the output sequence.
As you can see, Sample name, Locus name, and Paralog ranking are separated by double underscores (__). This is the reason why we don’t recommend using __ inside your sample names (see sample naming convention)
Created by Edgardo M. Ortiz (06.08.2021)
Last modified by Edgardo M. Ortiz (23.12.2024)