Input Preparation
At this point you should have de novo assemblies from your samples ready. However, Captus also gives you the flexibility of starting the analysis from this point by providing your own assemblies as FASTA files or complementing you newly aasembled samples with other assemblies (e.g. genomes or transcriptomes from GenBank). If you want to do so, please read the following note:
Reference target datasets formatting
Most importantly, in order to extract markers, the sequences in your reference target datasets have to follow a simple naming convention if you want to take advantage of using multiple reference sequences per locus and our paralog filtering method. When multiple reference sequences per locus are found in the reference dataset, Captus will decide during the extraction which of those references matches your assembly best based on similarity and total recovered length percentage.
Here is an example of a reference protein dataset that has two loci (called accD and cemA) with five reference sequences each (probably coming from different taxa to expand phylogenetic coverage). Coding sequences can be provided in either aminoacid or nucleotide. Miscellaneous DNA markers references can only contain nucleotide sequences.
>AA-S46062.1-accD [cluster_size=80]
MALQSLRGSMRSVVGKRICPLIEYAIFPPLPRIIVYASRRARMQRGNYSLIKKPKKVSTLRQYQSTKSPMYQSLQRICGVREWLNKYCMWKEVDEKDFG*
>AAZ94660.1-accD [cluster_size=17]
MEKRWLNSMLSKGELEYRCRLSKSINSLGPIESEGSIINNMNKNIPSHSDSYNSSYSTVDDLVGIRNFVSYDTFLVRDSNSSSYSIYLDIENQIFEIDN*
>ABH88096.1-accD [cluster_size=3]
MQKWRFNSMLLNRELEYGCEFKESLGPIENTSLNEEPKILSDIHKKINRWDDSDNSSYNSLDYLVGADNIQDFLSDKTFLVRDNKRNSYSIYLDIEKKT*
>ABW20568.1-accD [cluster_size=7]
MQNWINNSFQAEFEQESYFGSLGENSMNPRSGGDRYPEALIIRDITGETSAIYFDITDQILENDTHQTILASPIENDLWAEKDISIDIYRYINELIFYD*
>ACU46588.1-accD [cluster_size=1]
MAKYWFNLMLSYKMLSYNKLEHRCGLSKSMDNLNDLGHIGGNEELILNENDAKKNILGLENYNTHSINYLFDSRNIYNLIYNETFLVRNSNGYHYFVYF*
>QNK04966.1-cemA [cluster_size=1]
MKNKKAFIPLLHITFIVFLPWWIAFLFNKGLESWVINWWNTSKSEIFLNDIQEKNILEKFIELEELLLLDEMIKEYPET*
>QNP0849-5.1-cemA [cluster_size=4]
MTKKKAFTPIFYLSFLLFLPWWIDLLFNKCLRSWPTHWWNTRQSEMFLTTLQEKSLLEKFLELEEFLFLDKIIKKEFET*
>QNP08626.1-cemA [cluster_size=2]
MIKNKVFTPLFYLAFIVFLPWGIYFLLNKCMGSWTTNWWTTRESEILSTNINENSLLEKFIQFEEFLLLDEIIKKDTET*
>QNQ64689.1-cemA [cluster_size=9]
MAKNKICIPFISIVFLPWWISFLFKKDFESWVTNWWNTSKSEILLNDIQEKSILKTFIELEELFLLDEMLKEYPETRLQ*
>QPZ48083.1-cemA [cluster_size=7]
MAKKKAFISLIYLASIVFLPWWLSFTFNKSMESWVKNCWNTGPSENFLNDIEEKIIIKKFIELEELSLFDEILKDYTQD*
Let’s take a look at how the sequence names are formatted:
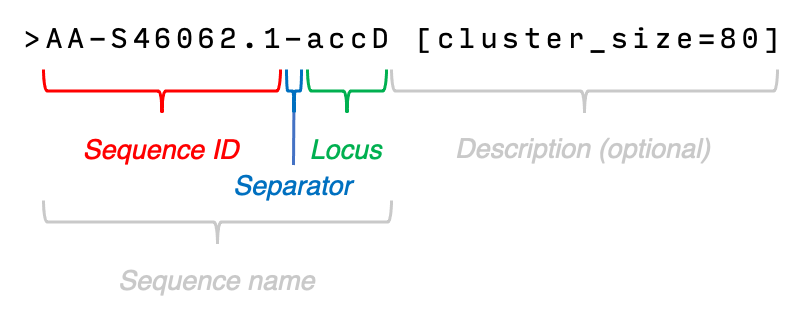
- The sequence name (any text found before the first space) can contain multiple
-characters, but only the last one will become the separator. - Any text found before the separator will be considered as sequence ID.
- Any text found after the the separator will become the locus name.
- Any text found after the first space is considered the description and this text is optional.
Angiosperms353 and HybPiper use this format, therefore, in order to mantain compatibility, we also used it to build our reference datasets for plastome proteins SeedPlantsPTD and mitochondrial proteins SeedPlantsMIT. At the same time it is also compatible with the references needed by other pipelines (e.g. SeCaPr) which are only capable of dealing with a single sequence per locus.
When the separator is not present (even in a single sequence in the whole reference dataset), the entire sequence name will be used as the locus name. This is fine when your reference dataset only contains a single sequence per locus for example, but if you intend to use the “muti-sequence per locus” format, please ensure that every single sequence contains a - as separator.
Created by Edgardo M. Ortiz (06.08.2021)
Last modified by Edgardo M. Ortiz (29.05.2021)