Basic Tutorial
This basic tutorial takes you through the minimal but most general workflow of the captus_assembly pipeline.
0. Preparation
Installation
To run this tutorial, you need to install the following programs on your system:
Captusand its dependencies (see Installation)IQ-TREEversion 2.0 or higher (see IQ-TREE documentation)
If conda command is available, you can take the easiest way.
Simply run the two commands below to set up and activate an environment:
conda create -n captus -c bioconda -c conda-forge captus iqtree
conda activate captus
Check if Captus is installed:
captus -h
If a help message shows up in your terminal, you are ready to go!
Getting data
Download this file (169 MB) and place it in a directory where you want to run this tutorial.
Then, run the following commands to unzip the archive.
cd <path/to/your/directory> # Replace with actual path
tar -zxvf 00_raw_reads.tar.gz && rm 00_raw_reads.tar.gz
Now you should have 00_raw_reads directory containing eight compressed FASTQ files:
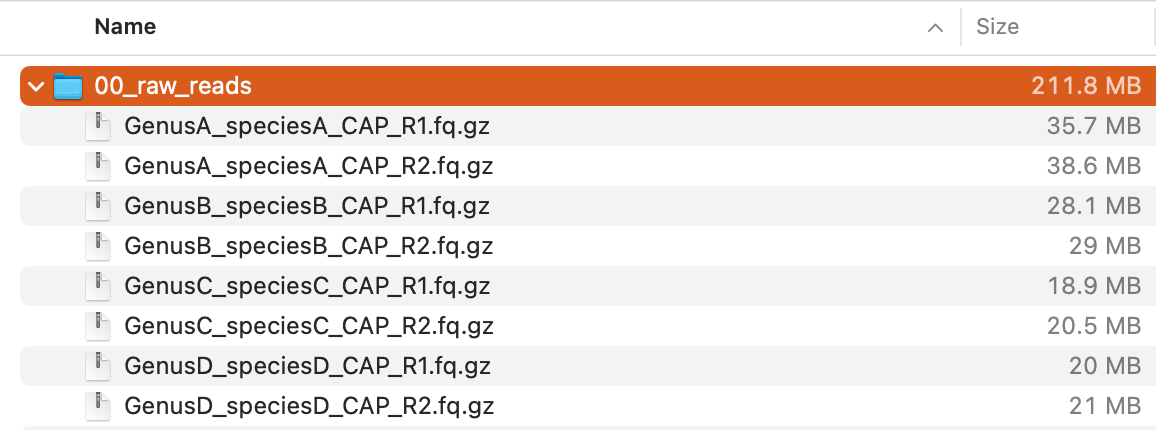 Those files are paired-end reads (
Those files are paired-end reads (R1 and R2) obtained from four plant species (GenusA_speciesA, GenusB_speciesB, GenusC_speciesC, GenusD_speciesD) by targeted-capture sequencing (CAP) of 353 loci highly conserved across angiosperms using the Angiosperms353 probe set (Johnson et al., 2018).
1. Cleaning Reads
Let’s start the analysis with cleaning the raw reads using the clean command.
The clean command trims adapter sequences and low-quality bases, and filters out reads with low average quality score.
When working with your own data, all FASTQ files must be named according to the naming convention.
Since Captus automatically recognizes sample name and library layout (single-end or paired-end) from the FASTQ file name, improper file naming may cause improper data processing.
00_raw_reads directory:
captus clean -r 00_raw_reads
-r: Path to directory containing FASTQ files.
If you want to customize the cleaning criteria, see Options.
This command will creat 01_clean_reads directory with the following structure:
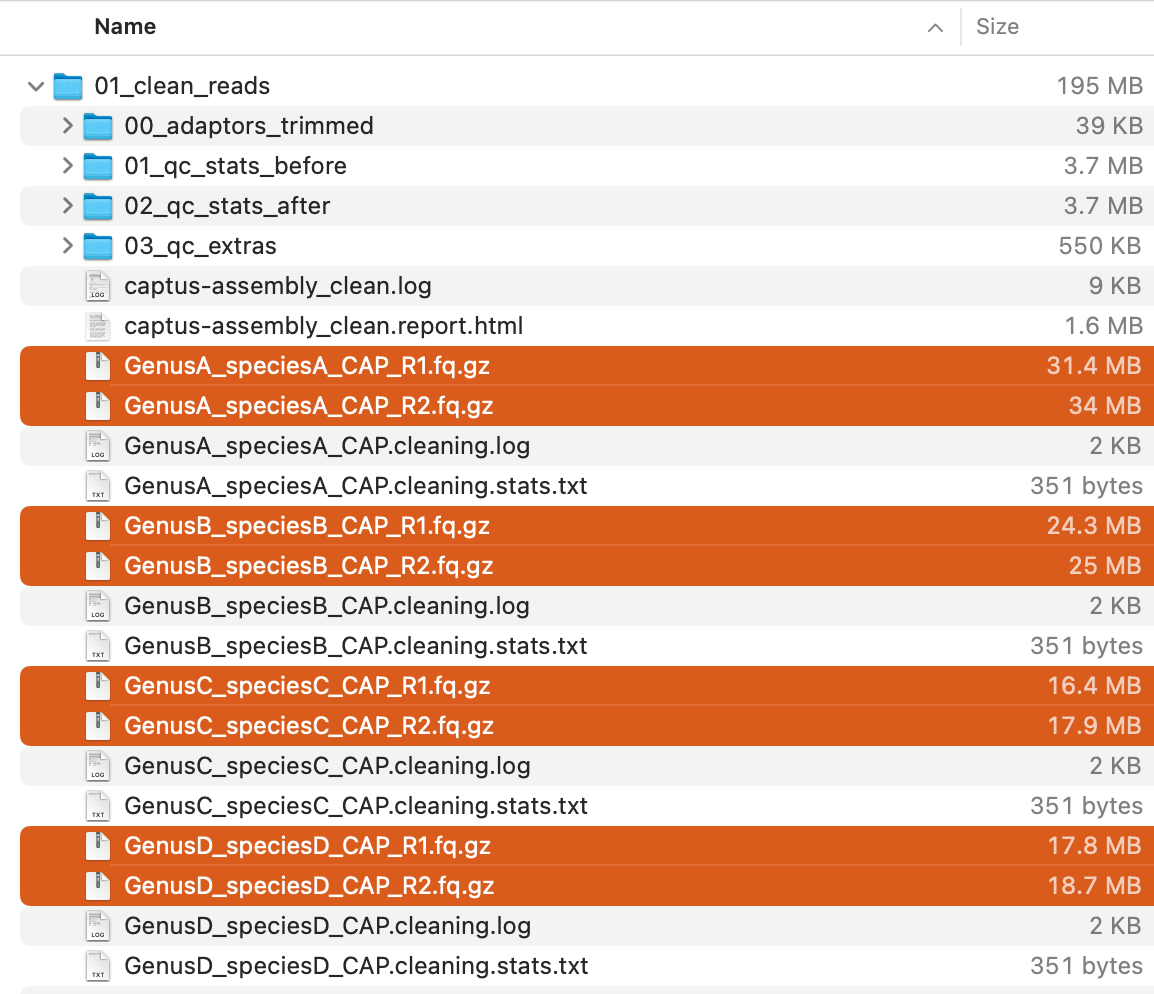
Of these, the compressed FASTQ files (highlighted in the image above) are the cleaned reads and will be used in the next step.
For descriptions of the other output files, see Output Files and HTML Report.
2. De Novo Assembly
Next, we would like to assemble the clean reads into contigs using the assemble command.
Run the following command to perform de novo assembly for all four samples with default settings optimized for targeted-capture and genome skimming data.
captus assemble -r 01_clean_reads
-r: Path to directory containing cleaned FASTQ files.
You can tune the assembler settings for your own data, see Options.
If you are worried about contaminations, consider to use the --max_contig_gc option.
The command will create a directory, 02_assemblies with the following structure:
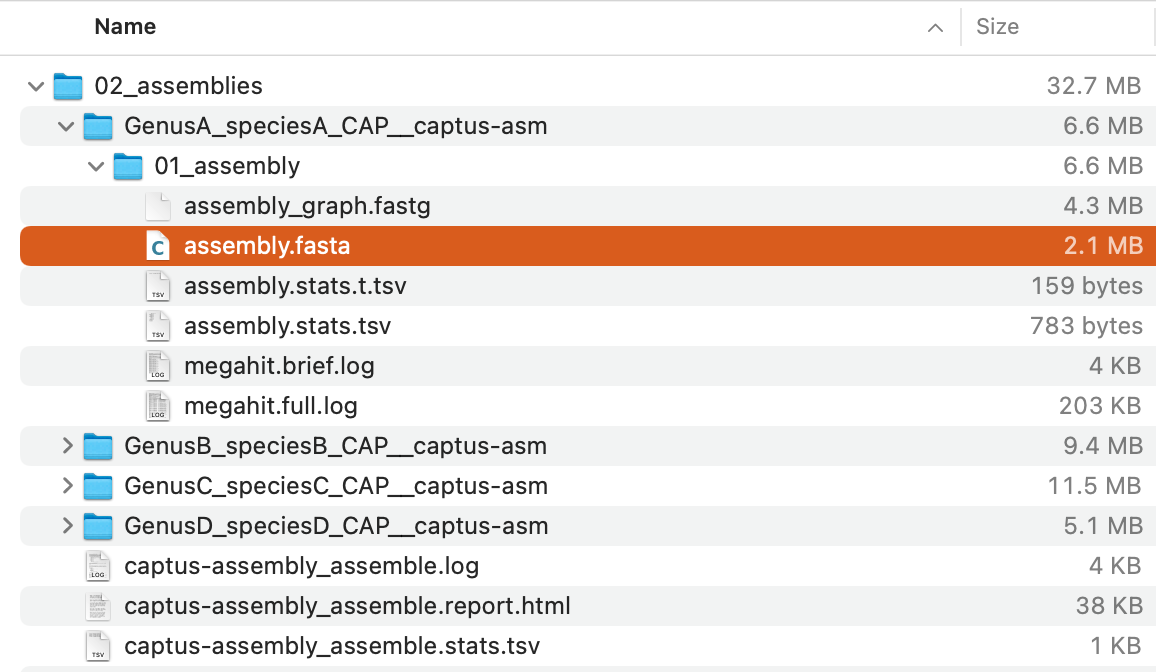
Of these, assembly.fasta in each directory (highlighted in the image above) is the assembled contigs.
For descriptions of the other output files, see Output Files and HTML Report.
3. Extracting Target Sequences
Now is the time of judgement.
We would like to extract the target gene sequences from the contigs assembled in the previous step using the extract command.
Run the following command to extract the sequences of all Angiosperms353 loci from all four samples:
captus extract -a 02_assemblies -n Angiosperms353
-a: Path to the output directory from theassemblecommand.-n: Path to a FASTA file of reference sequences. Here we use the built-inAngiosperms353reference dataset.
Captus can extract multiple marker types (nuclear proteins, plastidial proteins, mitochondrial proteins, and miscellaneous DNA markers) simultaneously.
For plastidial and mitochondrial proteins, Captus offers built-in reference datasets, SeedPlantsPTD and SeedPlantsMIT.
You can use them by simply adding -p SeedPlantsPTD and -m SeedPlantsMIT arguments to extract gene sequences from the organelle genomes of any flowering plants.
For descriptions of the other available options, see Options.
When providing your custom reference dataset, please make sure that all sequence names are formatted according to the Input Preparation.
For confirmation, Captus tells you in the log message how your reference dataset was recognized:
Nuclear proteins:
reference: Angiosperms353 /path/to/reference/Angiosperms353.FAA
reference info: 353 loci, 4,765 sequences (loci names found, detected multiple sequences per locus)
The command will creat 03_extractions directory with the following structure:
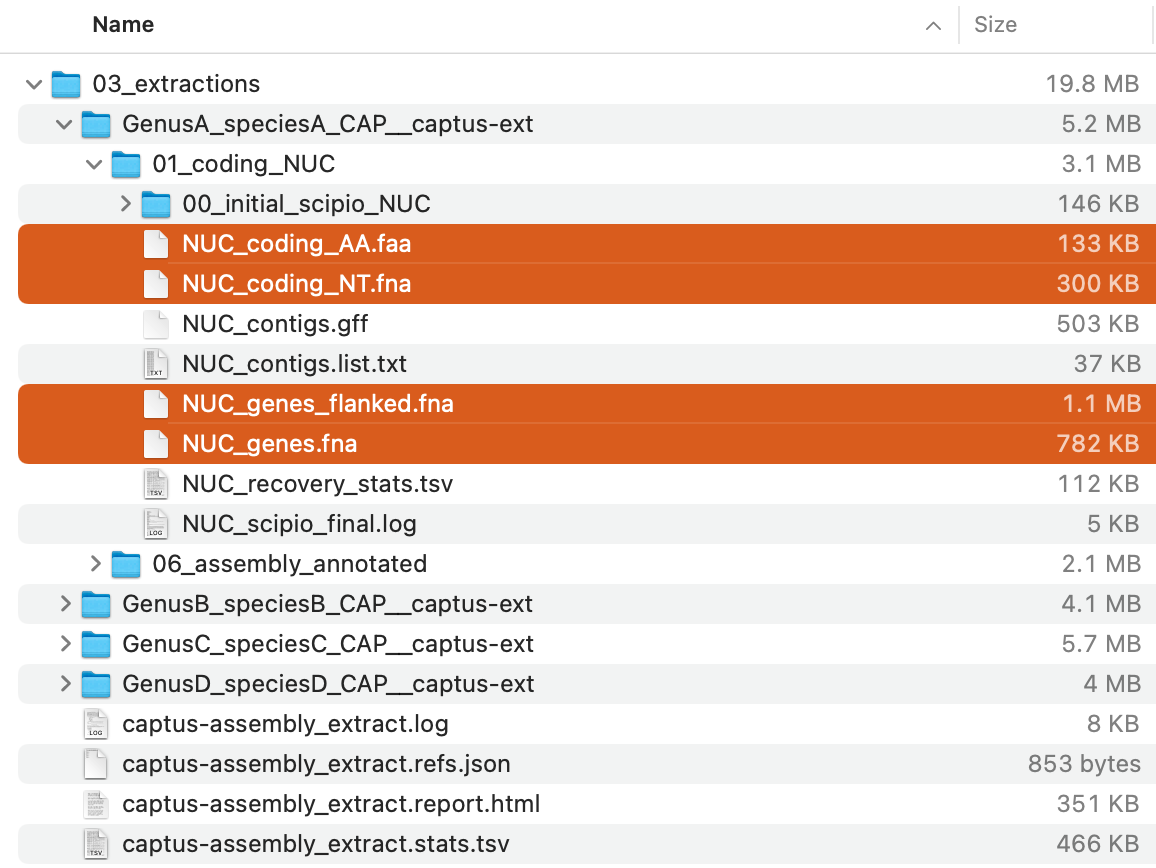
Of these, the FASTA files (*.faa and *.fna; highlighted in the image above) in each directory store the extracted sequences in each formats .
For descriptions of the other output files, see Output Files and HTML Report.
4. Multiple Sequence Alignment
The final step of the captus_assembly pipeline is to perform multiple sequence alignments on the sequences extracted from all samples for each locus using the align command to identify sequence variations among samples.
Run the following command to align the sequences extracted in the previous step, as well as to trim gappy ends and filter out paralogs from the alignments:
captus align -e 03_extractions
-e: Path to the output directory from theextractcommand.
For descriptions of the other available options, see Options.
The command will create 04_alignments directory with the following structure:
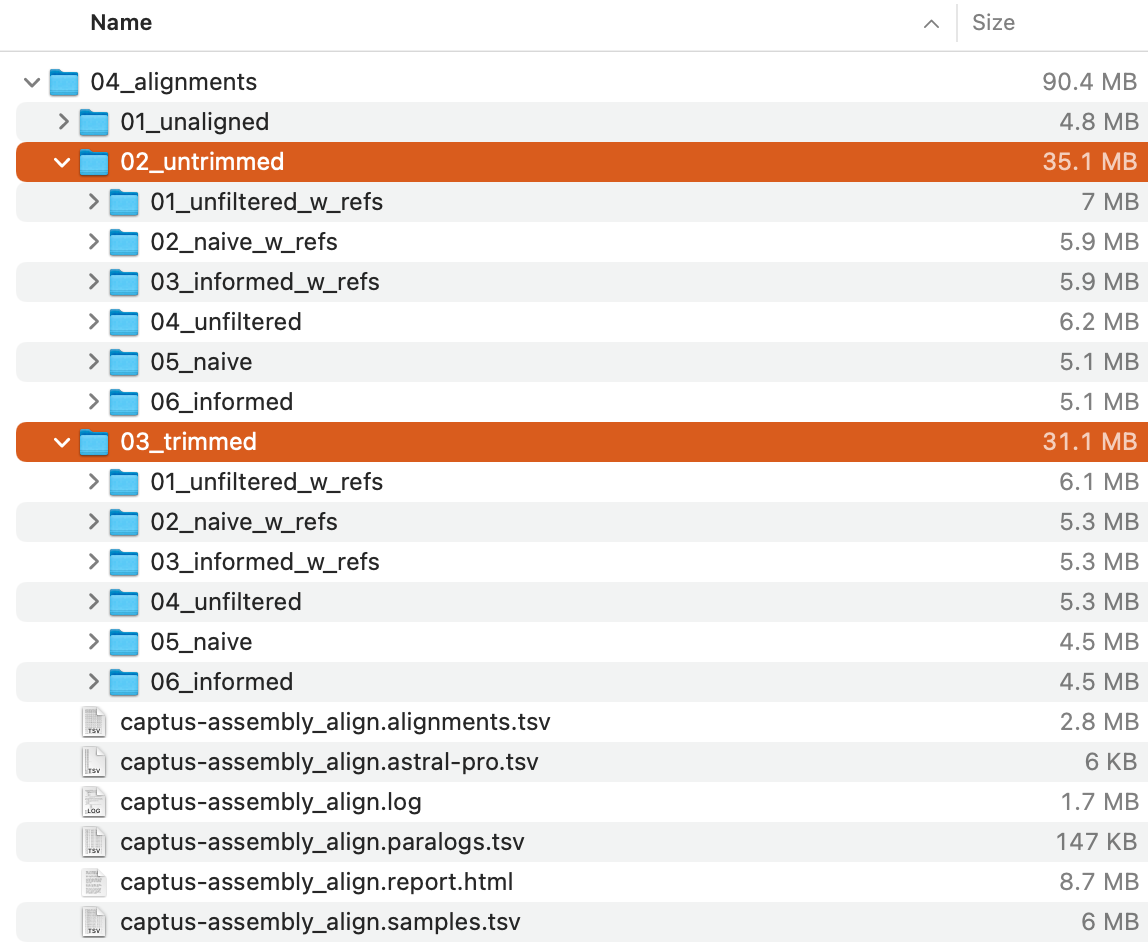 Of these,
Of these, 02_aligned_untrimmed and 03_aligned_trimmed directories (highlighted in the image above) store a series of untrimmed and trimmed alignments in multi-FASTA format, respectively.
Since the structure of the output directory is a bit complicated, we recommend that you take a look at Output Files and HTML Report.
5. Phylogenetic Inference
One of the most typical analyses after the captus_assembly pipeline would be phylogenetic tree inference.
Here we show a simple example of phylogenetic tree inference using IQ-TREE.
Run the following commands to infer a concatenation-based species tree using all loci with an edge-linked partition model:
mkdir 05_phylogeny && cd 05_phylogeny
iqtree -p ../04_alignments/03_trimmed/06_informed/01_coding_NUC/02_NT -pre concat -T AUTO
-p: NEXUS/RAxML partition file or path to a directory with alignments-pre: Prefix for output files-T: Number of cores/threads to use or AUTO-detect (default: 1)
For more practical uses of IQ-TREE, see IQ-TREE documentation.
The command will create the following files:
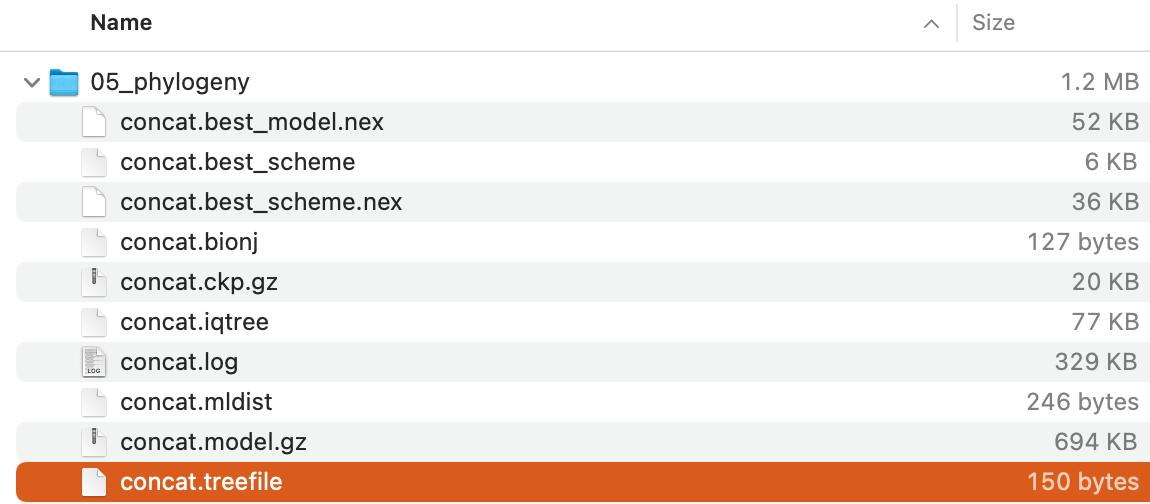 Of these,
Of these, concat.treefile is the file storing the maximum-likelihood tree in Newick format.
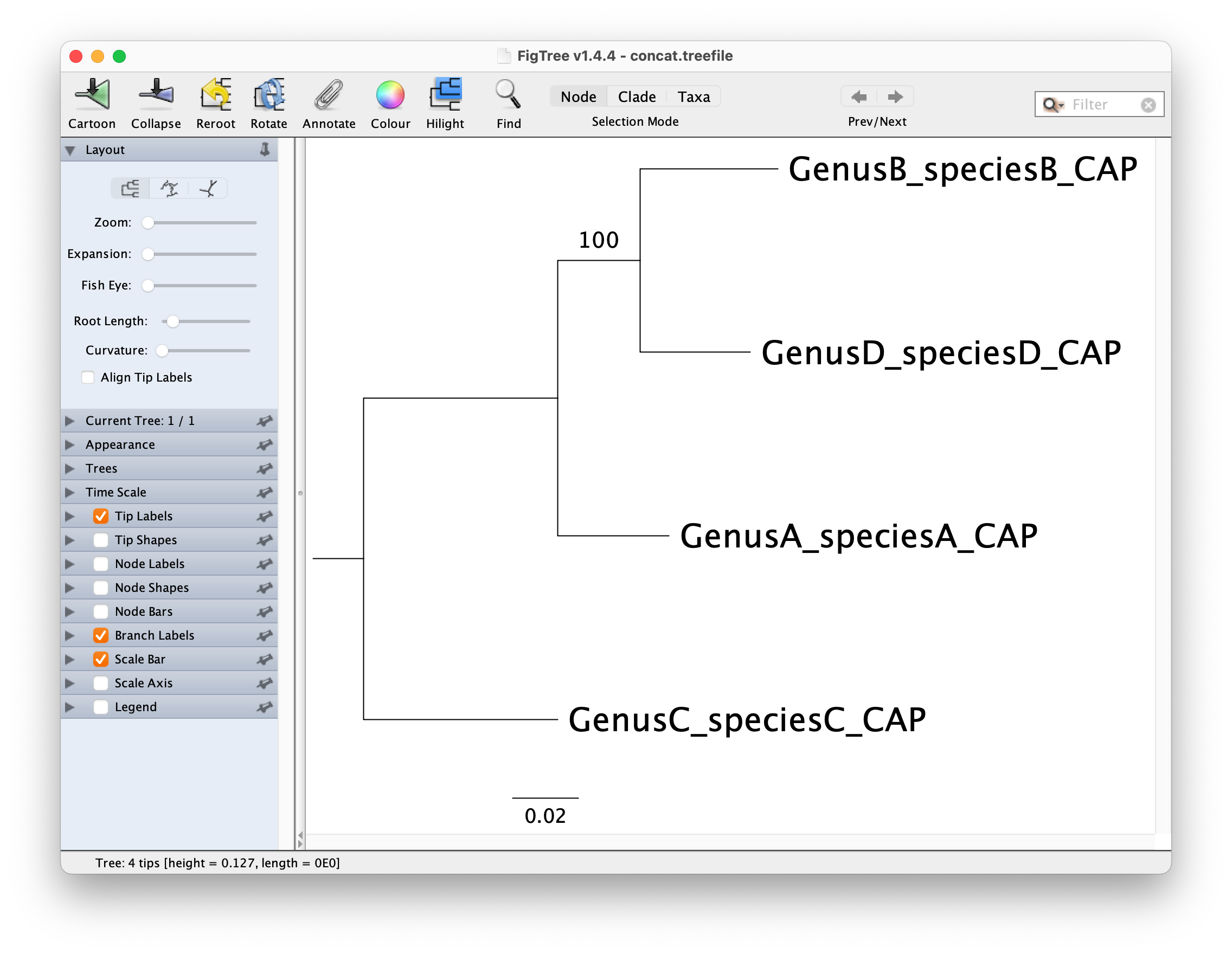
You can visualize this tree with a phylogenetic tree viewer such as FigTree or Dendroscope.
The maximum-likelihood tree opened in FigTree and rerooted on GenusC_speciesC_CAP should look like below:
 That’s all for the basic tutorial, but remember that this is a minimal usage of the
That’s all for the basic tutorial, but remember that this is a minimal usage of the captus_assembly pipeline.
To get the most out of this pipeline, such as integrating different data types at different processing steps or discovering new markers by clustering contigs, check out the Advanced Tutorial (currently under construction).
Created by Gentaro Shigita (01.10.2021)
Last modified by Gentaro Shigita (14.12.2024)