Output Files
Imagine we start with a directory called 00_raw_reads with the following content:
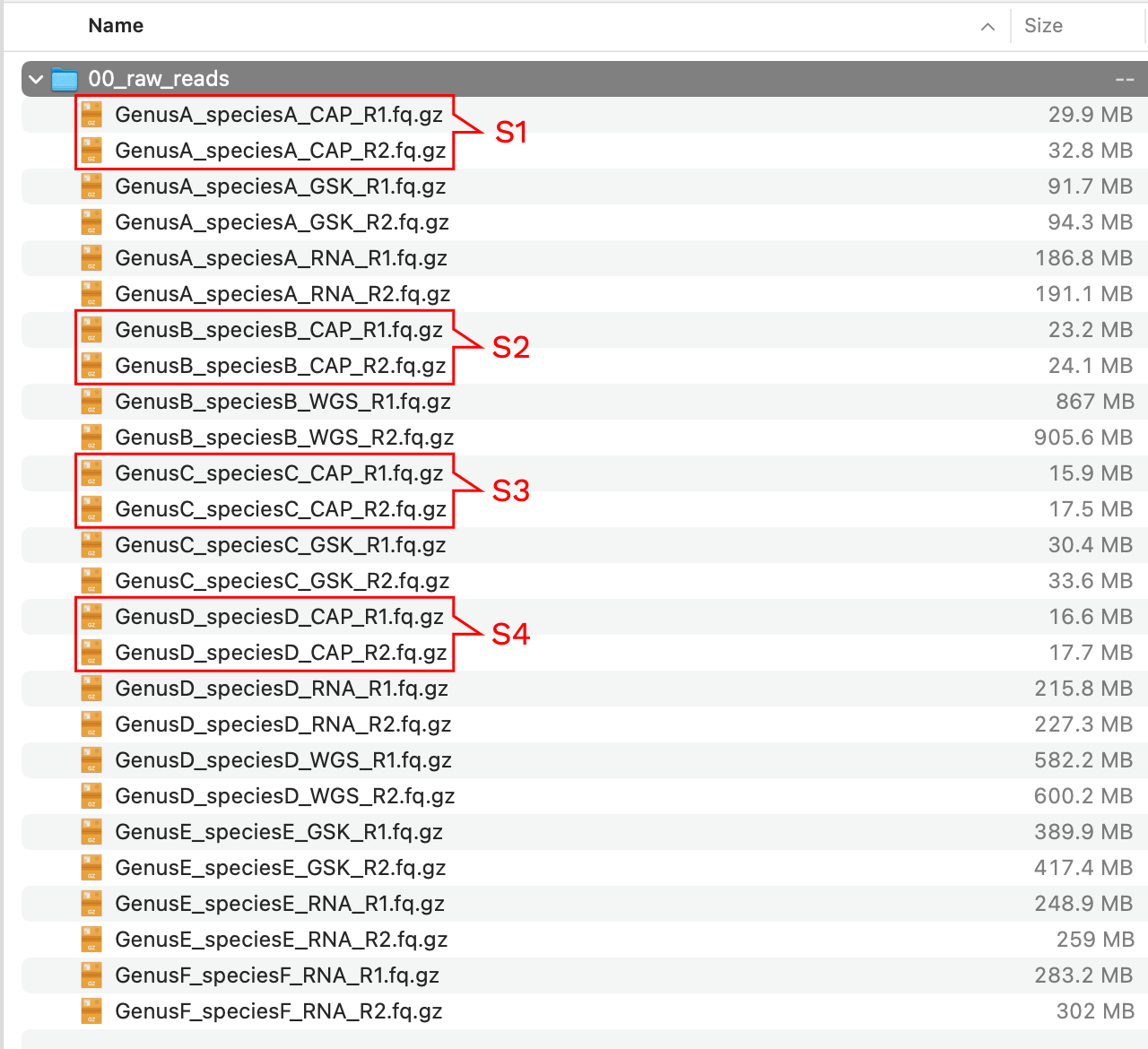
We have a samples with different data types, to distinguish them we added _CAP to the samples where hybridization-capture was used, _WGS for high-coverage whole genome sequencing, _RNA for RNA-Seq reads, and _GSK for genome skimming data (notice also the difference in file sizes). For this example, we only want to clean the samples in red rectangles corresponding to capture data. We run the following Captus command:
captus clean --reads ./00_raw_reads/*_CAP_R?.fq.gz
Notice we are using default settings, the only required argument is the location of the raw reads. The output was written to a new directory called 01_clean_reads. Let’s take a look at the contents:
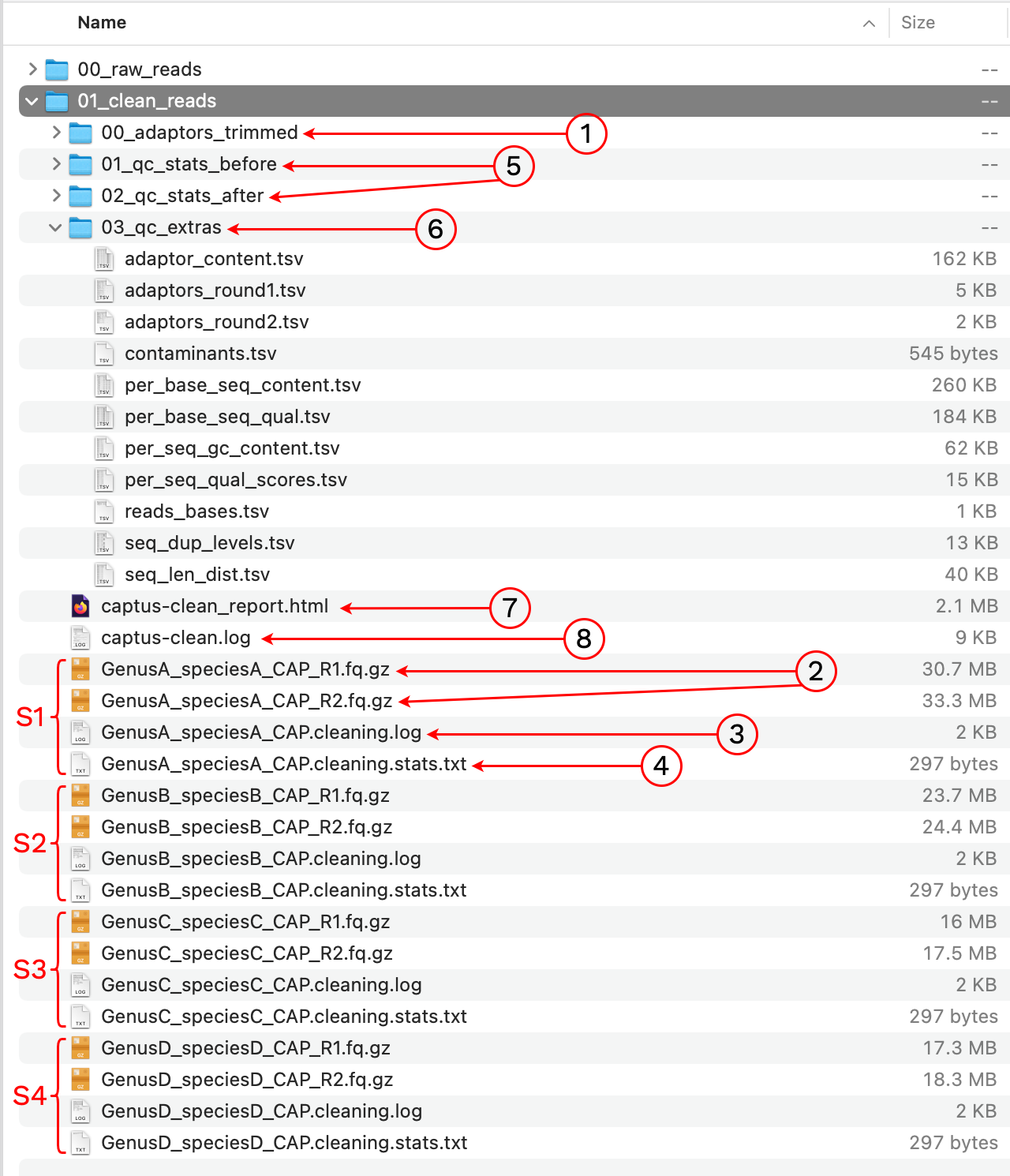
1. 00_adaptors_trimmed
This is an intermediate directory that contains the FASTQ files without adaptors, prior to quality-trimming and filtering. The directory also stores bbduk.sh commands and logs for the adaptor trimming stage. If the option --keep_all was enabled the FASTQs from this intermediate are kept after the run, otherwise they are deleted.
Example
[sample].round1.log
Captus' BBDuk Command for BOTH rounds:
bbduk.sh -Xmx8110m threads=8 ktrim=r minlength=21 interleaved=f trimpairsevenly=t trimbyoverlap=t overwrite=t ref=/software/GitHub/Captus/data/adaptors_combined.fasta in=/tutorial/00_raw_reads/GenusA_speciesA_CAP_R#.fq.gz out=stdout.fq ftr=0 k=21 mink=11 hdist=2 stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round1.stats.txt 2>/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.stdout1.log | bbduk.sh -Xmx8110m threads=8 ktrim=r minlength=21 interleaved=f trimpairsevenly=t trimbyoverlap=t overwrite=t ref=/software/GitHub/Captus/data/adaptors_combined.fasta in=stdin.fq out=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP_R#.fq.gz k=19 mink=9 hdist=1 stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round2.stats.txt 2>/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.stdout2.log
ROUND 1 LOG:
Executing jgi.BBDuk [-Xmx8110m, threads=8, ktrim=r, minlength=21, interleaved=f, trimpairsevenly=t, trimbyoverlap=t, overwrite=t, ref=/software/GitHub/Captus/data/adaptors_combined.fasta, in=/tutorial/00_raw_reads/GenusA_speciesA_CAP_R#.fq.gz, out=stdout.fq, ftr=0, k=21, mink=11, hdist=2, stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round1.stats.txt]
Version 38.95
Set threads to 8
Set INTERLEAVED to false
maskMiddle was disabled because useShortKmers=true
0.030 seconds.
Initial:
Memory: max=8503m, total=8503m, free=8478m, used=25m
Added 11201253 kmers; time: 3.185 seconds.
Memory: max=8503m, total=8503m, free=7967m, used=536m
Input is being processed as paired
Started output streams: 0.044 seconds.
Processing time: 8.051 seconds.
Input: 733778 reads 110800478 bases.
KTrimmed: 20580 reads (2.80%) 393486 bases (0.36%)
Trimmed by overlap: 1870 reads (0.25%) 10642 bases (0.01%)
Total Removed: 330 reads (0.04%) 404128 bases (0.36%)
Result: 733448 reads (99.96%) 110396350 bases (99.64%)
Time: 11.312 seconds.
Reads Processed: 733k 64.87k reads/sec
Bases Processed: 110m 9.79m bases/sec
[sample].round1.stats.txt
#File /tutorial/00_raw_reads/GenusA_speciesA_CAP_R1.fq.gz /tutorial/00_raw_reads/GenusA_speciesA_CAP_R2.fq.gz
#Total 733778
#Matched 11733 1.59898%
#Name Reads ReadsPct
Reverse_adaptor 1899 0.25880%
PhiX_read2_adaptor 1109 0.15114%
TruSeq_Adaptor_Index_1_6 1018 0.13873%
pcr_dimer 675 0.09199%
Forward_filter 608 0.08286%
Illumina 3p RNA Adaptor 595 0.08109%
I5_Nextera_Transposase_1 475 0.06473%
PhiX_read1_adaptor 468 0.06378%
Nextera_LMP_Read2_External_Adaptor 446 0.06078%
Reverse_filter 419 0.05710%
.
.
.
TruSeq_Adaptor_Index_9 1 0.00014%
[sample].round2.log
Captus' BBDuk Command for BOTH rounds:
bbduk.sh -Xmx8110m threads=8 ktrim=r minlength=21 interleaved=f trimpairsevenly=t trimbyoverlap=t overwrite=t ref=/software/GitHub/Captus/data/adaptors_combined.fasta in=/tutorial/00_raw_reads/GenusA_speciesA_CAP_R#.fq.gz out=stdout.fq ftr=0 k=21 mink=11 hdist=2 stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round1.stats.txt 2>/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.stdout1.log | bbduk.sh -Xmx8110m threads=8 ktrim=r minlength=21 interleaved=f trimpairsevenly=t trimbyoverlap=t overwrite=t ref=/software/GitHub/Captus/data/adaptors_combined.fasta in=stdin.fq out=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP_R#.fq.gz k=19 mink=9 hdist=1 stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round2.stats.txt 2>/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.stdout2.log
ROUND 2 LOG:
Executing jgi.BBDuk [-Xmx8110m, threads=8, ktrim=r, minlength=21, interleaved=f, trimpairsevenly=t, trimbyoverlap=t, overwrite=t, ref=/software/GitHub/Captus/data/adaptors_combined.fasta, in=stdin.fq, out=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP_R#.fq.gz, k=19, mink=9, hdist=1, stats=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP.round2.stats.txt]
Version 38.95
Set threads to 8
Set INTERLEAVED to false
maskMiddle was disabled because useShortKmers=true
Forcing interleaved input because paired output was specified for a single input file.
0.028 seconds.
Initial:
Memory: max=8503m, total=8503m, free=8478m, used=25m
Added 322384 kmers; time: 0.239 seconds.
Memory: max=8503m, total=8503m, free=8469m, used=34m
Input is being processed as paired
Started output streams: 0.056 seconds.
Processing time: 11.134 seconds.
Input: 733448 reads 110396350 bases.
KTrimmed: 10720 reads (1.46%) 103592 bases (0.09%)
Trimmed by overlap: 0 reads (0.00%) 0 bases (0.00%)
Total Removed: 18 reads (0.00%) 103592 bases (0.09%)
Result: 733430 reads (100.00%) 110292758 bases (99.91%)
Time: 11.447 seconds.
Reads Processed: 733k 64.07k reads/sec
Bases Processed: 110m 9.64m bases/sec
[sample].round2.stats.txt
#File stdin.fq
#Total 733448
#Matched 5386 0.73434%
#Name Reads ReadsPct
PhiX_read2_adaptor 781 0.10648%
Forward_filter 374 0.05099%
I5_Nextera_Transposase_1 373 0.05086%
Reverse_adaptor 367 0.05004%
RNA_Adaptor_(RA3)_part_#_15013207 361 0.04922%
Reverse_filter 327 0.04458%
PhiX_read1_adaptor 267 0.03640%
Stop_Oligo_(STP)_8 249 0.03395%
Nextera_LMP_Read2_External_Adaptor 215 0.02931%
Illumina 3p RNA Adaptor 190 0.02591%
.
.
.
I5_Primer_Nextera_XT_Index_Kit_v2_S520 1 0.00014%
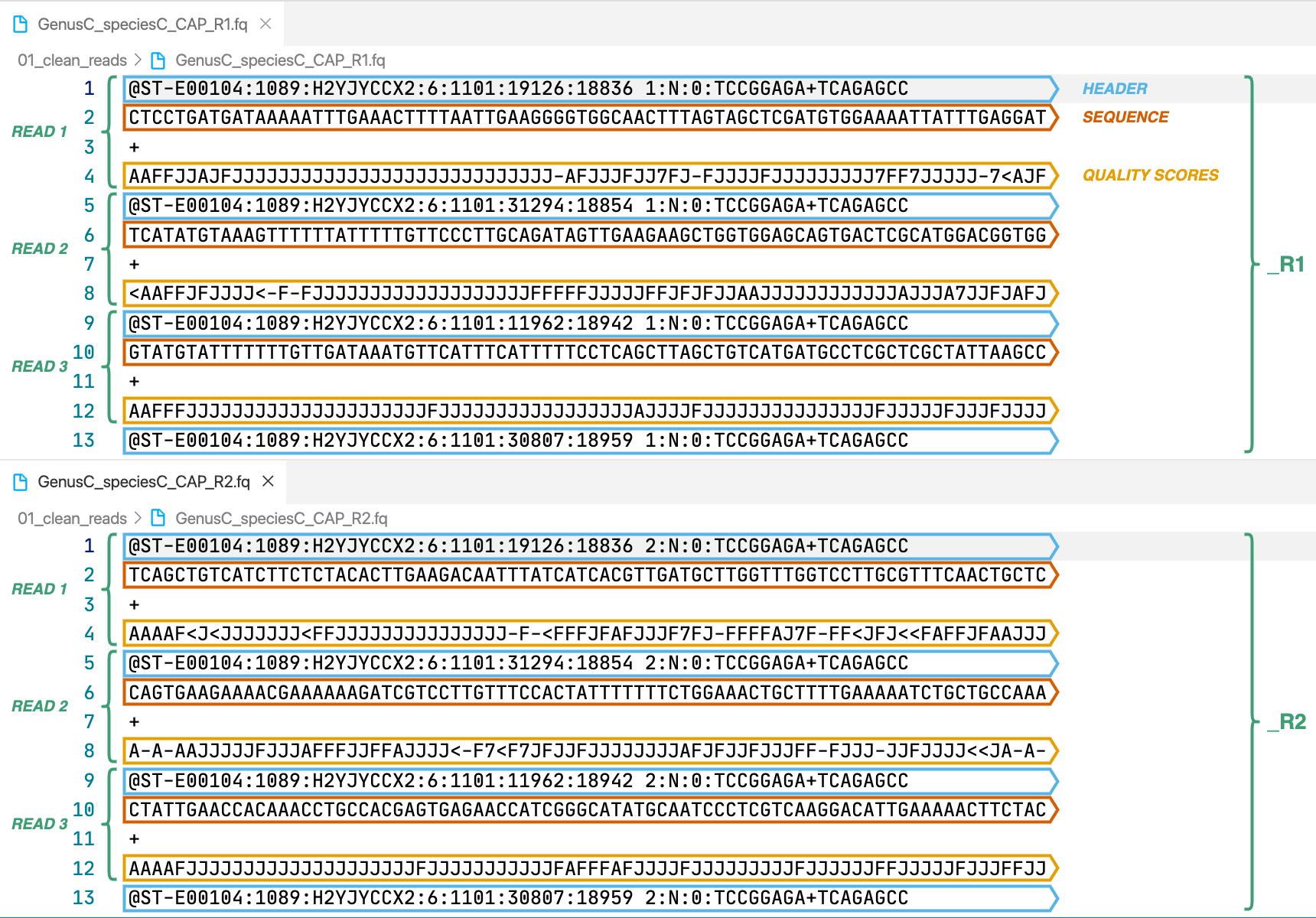
2. [sample]_R1.fq.gz, [sample]_R2.fq.gz
In case of paired-end input we will have a pair of files like in the image, the forward reads are indicated by _R1 and the reverse reads by _R2. Single-end input will only return forward reads. Wikipedia’s entry for the format describes it in more detail. These are the cleaned reads that will be used by the assemble module.
3. [sample].cleaning.log
This file contains the cleaning command used for bbduk.sh as well the data shown as screen output, this and other information is compiled in the Cleaning report.
Example
Captus' BBDuk Command:
bbduk.sh -Xmx16220m threads=8 in=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP_R#.fq.gz out=/tutorial/01_clean_reads/GenusA_speciesA_CAP_R#.fq.gz ref=/software/GitHub/Captus/data/phix174_ill.ref.fa.gz,/software/GitHub/Captus/data/sequencing_artifacts.fasta k=31 hdist=1 qtrim=lr trimq=13 maq=16 ftl=0 ftr=0 minlength=21 maxns=5 ziplevel=5 overwrite=t stats=/tutorial/01_clean_reads/GenusA_speciesA_CAP.cleaning.stats.txt 2>/tutorial/01_clean_reads/GenusA_speciesA_CAP.stdout.log
Executing jgi.BBDuk [-Xmx16220m, threads=8, in=/tutorial/01_clean_reads/00_adaptors_trimmed/GenusA_speciesA_CAP_R#.fq.gz, out=/tutorial/01_clean_reads/GenusA_speciesA_CAP_R#.fq.gz, ref=/software/GitHub/Captus/data/phix174_ill.ref.fa.gz,/software/GitHub/Captus/data/sequencing_artifacts.fasta, k=31, hdist=1, qtrim=lr, trimq=13, maq=16, ftl=0, ftr=0, minlength=21, maxns=5, ziplevel=5, overwrite=t, stats=/tutorial/01_clean_reads/GenusA_speciesA_CAP.cleaning.stats.txt]
Version 38.95
Set threads to 8
0.018 seconds.
Initial:
Memory: max=17007m, total=17007m, free=16987m, used=20m
Added 8403228 kmers; time: 1.021 seconds.
Memory: max=17007m, total=17007m, free=16612m, used=395m
Input is being processed as paired
Started output streams: 0.062 seconds.
Processing time: 3.655 seconds.
Input: 733430 reads 110292758 bases.
Contaminants: 0 reads (0.00%) 0 bases (0.00%)
QTrimmed: 127322 reads (17.36%) 515529 bases (0.47%)
Low quality discards: 13310 reads (1.81%) 1903218 bases (1.73%)
Total Removed: 13340 reads (1.82%) 2418747 bases (2.19%)
Result: 720090 reads (98.18%) 107874011 bases (97.81%)
Time: 4.753 seconds.
Reads Processed: 733k 154.32k reads/sec
Bases Processed: 110m 23.21m bases/sec
4. [sample].cleaning.stats.txt
List of contaminants found by bbduk.sh in the input reads, sorted by abundance.
Example
#File /tutorial/01_clean_reads/00_adaptors_trimmed/GenusX_speciesX_CAP_R1.fq.gz /tutorial/01_clean_reads/00_adaptors_trimmed/GenusX_speciesX_CAP_R2.fq.gz
#Total 60621406
#Matched 25 0.00004%
#Name Reads ReadsPct
gi|9626372|ref|NC_001422.1| Coliphage phiX174, complete genome 14 0.00002%
contam_111 8 0.00001%
contam_32 1 0.00000%
contam_76 1 0.00000%
contam_87 1 0.00000%
5. 01_qc_stats_before, 02_qc_stats_after
These directories contain the results from either Falco or FastQC, organized in a subdirectory per FASTQ file analyzed.
This directory contains all the tab-separated-values tables needed to build the Cleaning report. We provide them separately to allow the user more detailed analyses.
List of tables produced
| Table |
Description |
| adaptor_content.tsv |
Adaptor content percentages, parsed from Falco’s orFastQC’s output |
| adaptors_round1.tsv |
Reads/bases after first round of adaptor removal, parsed from bbduk.sh’s logs |
| adaptors_round2.tsv |
Reads/bases after second round of adaptor removal, parsed from bbduk.sh’s logs |
| contaminants.tsv |
Contaminant content, compiled from bbduk.sh’s logs |
| per_base_seq_content.tsv |
Per base sequence content, parsed from Falco’s orFastQC’s output |
| per_base_seq_qual.tsv |
Per base sequence quality, parsed from Falco’s orFastQC’s output |
| per_seq_gc_content.tsv |
GC content per sequence, parsed from Falco’s orFastQC’s output |
| per_seq_qual_scores.tsv |
Per sequence quality scores, parsed from Falco’s orFastQC’s output |
| reads_bases.tsv |
Reads/bases after quality filtering and contaminant removal, parsed from bbduk.sh’s logs |
| seq_dup_levels.tsv |
Sequence duplication levels, parsed from Falco’s orFastQC’s output |
| seq_len_dist.tsv |
Sequence length distribution, parsed from Falco’s orFastQC’s output |
7. captus-clean_report.html
This is the final Cleaning report, summarizing statistics across all samples analyzed.
8. captus-clean.log
This is the log from Captus, it contains the command used and all the information shown during the run. If the option --show_less was enabled, the log will also contain all the extra detailed information that was hidden during the run.
Created by Edgardo M. Ortiz (06.08.2021)
Last modified by Edgardo M. Ortiz (23.12.2024)